
Mieloma múltiple asociado con hiperbetaglobulinemia en paciente cirrótico. Reporte de caso
Multiple Myeloma Associated with Hyperbetaglobulinemia in a Cirrhotic Patient. Case Report
Mieloma múltiplo associado a hiperbetaglobulinemia em paciente cirrótico. Reporte de caso
Mieloma múltiple asociado con hiperbetaglobulinemia en paciente cirrótico. Reporte de caso
Revista Ciencias de la Salud, vol. 17, núm. 3, 2019
Universidad del Rosario
Recibido: 25 Octubre 2018
Aceptado: 22 Abril 2019
Información adicional
Para citar este artículo: Ramírez Vargas R, Vera Marmanillo V, Velazque Rojas L, Pari Caller CG. Mieloma múltiple asociado con hiperbetaglobulinemia en paciente cirrótico. Reporte de caso. Rev Cienc Salud. 2019;17(3):132-140. Doi: https://doi.org/10.12804/revistas.urosario.edu.co/revsalud/a.8370
Resumen: Introducción: el mieloma múltiple es una enfermedad hematooncológica de origen clonal, caracterizada por la producción anormal de células plasmáticas que producen un reemplazo progresivo de otras líneas hematopoyéticas. El 95% de los casos condiciona el aumento de la producción de inmunoglobulinas defectuosas, predominantemente IgG e IgA, detectadas en suero con pico monoclonal, definido por un deterioro progresivo de la función renal aguda, anemia, citopenias, dolor óseo agudo o crónico, fracturas patológicas, trastornos endocrinos recientes y, como en el caso de estudio, asociado con cirrosis hepática. Presentación del caso: paciente masculino de 49 años, con cefalea, dolor abdominal, torácico, evacuaciones melénicas, disnea y signos de ascitis, con un tiempo de enfermedad de tres meses, con historia de dolor lumbosacro de tres años antes de su ingreso. Los exámenes auxiliares mostraron pancitopenia, perfil de coagulación alterado, función renal alterada, inversión de albúmina-globulina, hiperuricemia, ascitis, lesiones óseas líticas, hipergammapatía monoclonal de tipo IgA relacionado con hiperbetaglobulinemia. Los exámenes específicos dieron como diagnóstico mieloma múltiple IgA EC IIIB más cirrosis hepática. Se presenta este caso como inusual por elevación de proteínas en suero, tanto beta como gamma, producto de dos patologías concomitantes. Conclusión: paciente con dos enfermedades simultáneas, en ambos casos con elevación de globulinas, lo que podría confundir el diagnóstico; el estado de inmunosupresión mejora con la administración de inmunoglobulinas y de tratamiento para la enfermedad base.
Palabras clave mieloma múltiple, enfermedad hematooncológica, IgG, IgA.
Abstract: Introduction: The multiple myeloma is a hemato-oncological disease of clonal origin, characterized by the abnormal production of plasma cells that produce a progressive replacement of the other hematopoietic lines in the bone marrow. In 95% of the cases, the condition showed an increase in the production of defective immunoglobulins, predominantly IgG and IgA detected in serum with monoclonal peak, characterized by progressive deterioration of acute renal function, anemia, cytopenias, acute or chronic bone pain, pathological fractures, and recent endocrine disorders. The present case associated with hepatic cirrhosis. Case presentation: A 49-year-old male patient with headache, abdominal pain, thoracic, manes, dyspnea and signs of ascites with a 3-month disease time, and lumbosacral pain history of 3 years before admission. Auxiliary examinations showed pancytopenia, altered coagulation profile, impaired renal function, albumin-globulin inversion, hyperuricemia, ascites, lytic bone lesions, and monoclonal hypergammapathy of IgA type associated with hyperbetaglobulinemia. The specific exams gave as diagnosis multiple IgA EC IIIB myeloma, plus liver cirrhosis, this case is presented as unusual by elevation of serum proteins in both beta and gamma product of two concomitant pathologies. Conclusion: A patient with two simultaneous diseases, both cases with globulin elevation that could confuse the diagnosis, whos state of immunosuppression improved with the administration of immunoglobulins and the treatment for the base disease.
Keywords: Multiple myeloma, hemato-oncological disease, IgG, IgA.
Resumo: Introdução: o mieloma múltiplo é um doença hemato-oncológica de origem clonal, caracterizada pela produção anormal de células plasmáticas que produzem uma substituição progressiva de outras linhas hematopoiéticas. O 95% dos casos, condiciona o aumento da produção de imunoglobulinas defeituosas, predominantemente IgG e IgA detectadas em soro com pico monoclonal, caracterizada por um deterioro progressivo da função renal aguda, anemia, citopenias, dor óssea aguda ou crónica, fraturas patológicas, transtornos endócrinos recentes e no presente caso associado à cirrose hepática. Apresentação do caso: paciente masculino de 49 anos com cefaleia, dor abdominal, torácica, melena, dispneia e signos de ascite com um tempo de doença de 3 meses, com história de dor lombossacral 3 anos antes de seu ingresso, os exames auxiliares mostraram pancitopenia, perfil de coagulação alterada, função renal alterada, investimento de albumina-globulina, hiperuricemia, ascite, lesões ósseas líticas, hipergamapatia monoclonal de tipo IgA associado a hiperbetaglobulinemia. Os exames específicos deram como diagnóstico mieloma múltiplo IgA EC IIIB mais cirrose hepática, se apresenta este caso como inusitada por elevação de proteínas em soro tanto beta quanto gama produto de duas patologias concomitantes. Conclusões: paciente com duas doenças simultâneas em ambos os casos com elevação de globulinas que poderiam confundir o diagnóstico e pelo estado de imunossupressão melhora com a administração de imunoglobulinas e tratamento para a doença base.
Palavras-chave: mieloma múltiplo, doença hemato-oncológica, IgG, IgA.
Introducción
El mieloma múltiple o enfermedad de Kahler es una hemopatía maligna caracterizada por una proliferación plasmocítica monoclonal que invade la médula ósea. Ocupa el segundo lugar entre las hemopatías en términos de prevalencia (1). El mieloma se caracteriza por la presencia de un pico estrecho, la mayoría de las veces en la zona de las gammaglobulinas, en el caso de los mielomas secretores; y por una hipogammaglobulinemia con una proteinuria de Bence-Jones en los mielomas de cadenas ligeras (1). A medida que las células cancerosas se multiplican, pueden causar dolor y destrucción de los huesos; si se afectan los de la columna, pueden ejercer presión sobre los nervios, lo que provoca dolor y parestesias. Ante un paciente de edad avanzada, con dolor intenso óseo no asociado con traumatismo, que no mejora con tratamiento, se debe realizar diagnóstico diferencial de mieloma múltiple (2, 3).
El mieloma múltiple representa el 15% de las neoplasias hematológicas, y su incidencia ha venido en ascenso constante en los últimos 30 años al mejorar las técnicas diagnósticas y al aumentar la esperanza de vida en la población general (3, 4). La incidencia se incrementa en forma progresiva con la edad, con un pico máximo entre los 50 y los 60 años, siendo solo el 2 % de los casos correspondientes a pacientes menores de 40 años, predomina en el género masculino y en la raza negra en una relación de 2:1 (4, 5).
Las manifestaciones clínicas más comunes están relacionadas con el daño de órgano blanco, presentadas en forma de anemia o pancitopenia, dolor óseo como síntoma común en el 70% de los casos y fracturas patológicas. Suelen encontrarse otras expresiones derivadas de la hipercalcemia por daño óseo o asociadas con la insuficiencia renal; así mismo, el hallazgo de más de 30 gramos por litro de proteína monoclonal, más del 10 % de plasmocitos en la médula ósea y el compromiso de dos órganos blancos en un año manifestado por hipercalcemia, daño renal, anemia, lesiones óseas, manifestaciones derivadas de hiperviscosidad e infecciones bacterianas recurrentes son considerados criterios clínicos para el diagnóstico oportuno de la enfermedad (5).
La estadificación de los pacientes con mieloma múltiple cumple un papel importante para el tratamiento y pronóstico de la enfermedad, se ha realizado tradicionalmente según la clasificación propuesta por Durie y Salmón; sin embargo, el sistema de estadificación recientemente propuesto por el International Staging System divide los pacientes en tres grupos de riesgo con base en los niveles de β2-microglobulina y de albúmina (6).
El mieloma múltiple, a pesar de ser considerada una enfermedad incurable, plantea como tratamiento la utilización de quimioterapia en altas dosis, seguida del trasplante autólogo de células progenitoras. Este procedimiento es avalado, en la actualidad, como el tratamiento estándar para los pacientes menores de 55 años (7). Para los pacientes mayores de 55 años, el uso de fármacos como la talidomida, en combinación con los esteroides y otros medicamentos como los inhibidores del proteosoma (bortezomib), ha demostrado actividad sinérgica en el tratamiento (8).
Las células idiotípicas del mieloma se pueden encontrar en la sangre de los pacientes en todos los estadios de la enfermedad. Por esta razón, se debe considerar el tratamiento sistémico para todos los pacientes con neoplasias de células plasmáticas sintomáticas (9, 10).
El reto más importante se justifica en sospechar, evaluar, diagnosticar y separar al grupo de pacientes asintomáticos estables, que no necesitan tratamiento, de los pacientes de mieloma sintomático que evolucionan desfavorablemente, los cuales deben ser tratados de inmediato.
A continuación, se expone el caso de un paciente con mieloma múltiple asociado con cirrosis hepática, que representó un reto en el diagnóstico y tratamiento.
Presentación del caso
Paciente masculino de 49 años, natural y procedente de Quillabamba (Cusco, Perú), que ingresa al servicio de emergencia con síntomas inespecíficos de tres años de evolución, caracterizados por presentar dolor a nivel lumbosacro, que cedían con analgésicos, intensificados durante los últimos tres meses. El cuadro clínico manifestado a su ingreso fue dolor abdominal localizado en hipocondrio derecho, dolor torácico, dolor lumbar, evacuaciones melénicas, hematemesis y disnea. Como antecedentes relevantes: alcoholismo desde hace 23 años, hospitalizaciones en dos oportunidades por dolor óseo inespecífico; al examen físico se mostró hemodinámicamente estable, despierto, lucido, piel pálida, tórax con murmullo vesicular conservado disminuido en ambas bases, ruidos cardíacos rítmicos normofonéticos, abdomen distendido, ruidos hidroaéreos disminuidos, matidez desplazable, dolor difuso a la palpación, onda ascítica (+), extremidades hipotróficas, reducción de la motilidad, dolor a la presión a nivel de la columna dorso-lumbosacra (Dandy +), despierto, orientado en tiempo, espacio y persona.
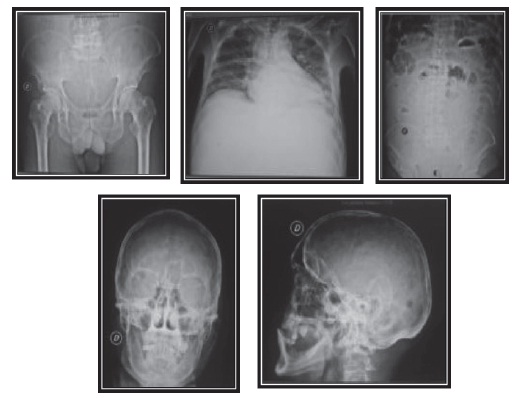
La analítica muestra: leucopenia 2.660, hemoglobina del 4,9%, hematocrito al 14,4%, plaquetas 58 000 mm3, glucosa 85 mg, urea 52 mg, creatinina 2 mg, proteínas totales 11,4 g/dl, albúmina 2,3 g/dl, globulina 9,10 g/dl, bilirrubina total 0,36, dhl 64 U/l, tgo 30 U/l, tgp 11 U/l, examen de orina patológico, inr: 3,15, ecografía abdominal: cirrosis hepática con ascitis, endoscopia alta normal, estudio de líquido ascítico pap (-), block cell (-), bk (-), gram y cultivo (-), adenosina de amina (-), calcio sérico 8,8, vsg: 84 mm/h, proteinuria en orina de 24 horas 0,38 g/24 h, proteína de Bence-Jones negativo. El resultado de la prueba β2-microglobulina sérica se muestra elevada (6.0 mg/l). Al rastreo óseo de pelvis, abdomen, tórax y cráneo, se observan lesiones de tipo sacabocados (figura 1), por lo que se realiza biopsia de hueso con resultado de plasmocitoma (figura 2).
Electroforesis de proteínas en suero: hipoalbuminemia 1,88 g/dt, globulinas alfa-1 0,35 g/dt, alfa-2 0,98 g/dt y beta con hiperbetaglobulinemia 6,34 g/dt (figura 3). Después de la elaboración completa y evolución de los cuadros clínicos, se llegó a diagnóstico definitivo de mieloma múltiple IgA EC IIIB asociado con cirrosis hepática, resultado que permitió planificar el tratamiento basado en talidomida, ciclofosfamida, prednisona, sintomáticos y gammaglobulinas.
Discusión
El mieloma múltiple es una enfermedad de predominio en mayores de 50 años, pero también se puede presentar raramente en las primeras cuatro décadas de vida. Respecto al paciente de 49 años con antecedente de alcoholismo crónico de más de 20 años y con un cuadro de falla renal aguda, trastorno hematológico y síndrome ascítico, se sospecha rápidamente un cuadro cirrótico descompensado; pero, por el antecedente importante de presentar dolor óseo, más hospitalizaciones anteriores con el cuadro de dolor y observar la inversión albúmina-globulina marcada, se planteó la posibilidad de una gammapatía monoclonal, que, asociada con el estudio de imágenes donde se evidencian lesiones líticas en cráneo, pelvis, columna (figura 1), y vsg incrementado, orientó al diagnóstico de mieloma múltiple en paciente con cirrosis hepática. Por tal motivo, se efectuaron los exámenes específicos respectivos: biopsia de hueso, dosaje de inmunoglobulinas, proteinograma electroforético, β2-microglobulina, proteína de Bence-Jones. Así mismo, se pensó en una neoplasia cuando el valor de las alfa-2 globulinas, sumado al de las betaglobulinas, fue superior al doble del de la gammaglobulina. Con los anteriores criterios mencionados, se concluyó que el diagnóstico definitivo era mieloma múltiple IgA EC IIIB + cirrosis hepática.
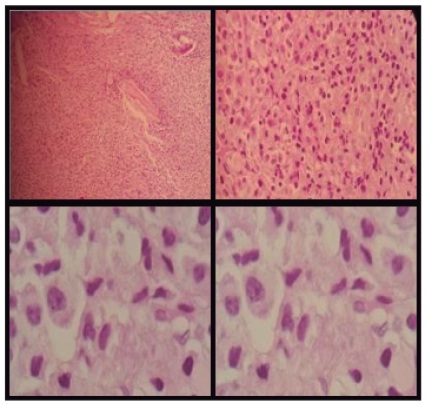
No obstante, la pancitopenia que presenta este paciente llevó a pensar que la causa era el mieloma múltiple que se produce por la proliferación de células plasmáticas en la médula ósea, por infiltración o reemplazo de la médula ósea; sin embargo, al estar asociado con cirrosis hepática, esta podría deberse a la hipertensión portal que conduce a la pancitopenia por hiperesplenismo, con lo que se concluye que la pancitopenia obedecía a una causa tanto central como periférica.
Se inició el tratamiento con talidomida, ciclofosfamida y corticoides. Idealmente el manejo debería ser con bortezomib, corticoides o lenalidomida, corticoides o bortezomib, talidomida, corticoides, pero no existe la disponibilidad de estos en la institución. Por otro lado, existe una controversia por tratarse de paciente cirrótico. Se administraron gammaglobulinas para mejorar el estado inmunológico, en el cual se evidenció una mejoría en curso de evolución del paciente, clínica y analíticamente. A la semana de culminada la administración de las gammaglobulinas y con aplicación de antibioticoterapia y tratamiento de soporte para la cirrosis hepática, al paciente se le dio de alta del establecimiento, debiendo continuar con los controles por consultorio externo.
El diagnóstico precoz o sospecha clínica de esta patología de identificación poco común en medicina general, y con presencia de la tríada clásica: anemia, falla renal y dolor óseo, tiene impacto en cuanto a pronóstico y respuesta al tratamiento, lo que optimiza la calidad de vida del paciente y años de sobrevida.
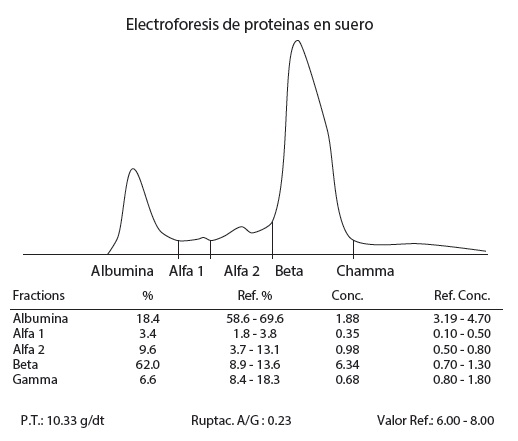
Es importante observar que este paciente presenta una elevación de la fracción beta en el proteinograma electroforético que no es frecuente encontrar en el mieloma múltiple, por lo cual es relevante el antecedente de cirrosis hepática, lo que permite llevar a cabo más estudios de asociación entre mieloma múltiple y otras enfermedades que muestren estas características.
Recomendaciones
El tratamiento de pacientes con este tipo de neoplasias ha sido enfocado a la restauración del daño, debido a que se carece de programas de tipo preventivo, por lo que resulta urgente evaluar alternativas concretas tanto de prevención primaria como de detección oportuna, modificando los factores de riesgo.
Es necesario optimizar el sistema de registro de morbilidad y mortalidad para las enfermedades hematooncológicas, ya que el subregistro se incrementa cuando los enfermos son diagnosticados en estadios avanzados, lo que provoca la defunción intradomiciliaria, sin atención previa. La mejoría en los registros facilitará la vigilancia epidemiológica de las enfermedades hematooncológicas, la evaluación del impacto de programas específicos para la prevención, diagnóstico temprano, control de enfermedad, tratamiento y sobrevida.
Contribución de los autores
Renán Ramírez Vargas: autor principal. Identificación del caso, diagnóstico, control del paciente. Búsqueda de imágenes para el manuscrito, revisión de la historia clínica, escritura del caso y edición del manuscrito. Verónika Vera Marmanillo: selección del caso e imágenes para el manuscrito, búsqueda de la literatura, revisión de la historia clínica, determinación de los análisis en laboratorio. Lida Velazque Rojas y Charlye Gyorgy Pari Caller: identificación del caso y selección de imágenes para el manuscrito, revisión de la historia clínica y literatura, redacción y edición del manuscrito.
Conflicto de intereses
Ninguno declarado.
Referencias
1. Charlot-Lambrecht I, Salmon J-H, Gagneux-Lemoussu L, Brochot P, Eschard J-P. Mieloma múltiple. emc - Apar Locomot. 2012;45(1):1-13. Doi: 10.1016/S1286-935X (12)60820-X
2. Álvarez-Cordovés MM, Mirpuri-Mirpuri PG, Pérez-Monje A. Diagnóstico de mieloma múltiple en atención primaria. Sospecha ante una historia clínica adecuada. Semer - Med Fam. 2013;39(6):e21-4. Doi: 10.1016/J.SEMERG.2012.05.006
3. Martínez-López J, Blade J, Mateos M-V, Grande C, Alegre A, García-Laraña J, et al. Long-term prognostic significance of response in multiple myeloma after stem cell transplantation. Blood. 2011;118(3):529-34. Doi: 10.1182/blood-2011-01-332320
4. Rajkumar SV, Landgren O, Mateos M-V, Kyle R, Remstein E, Therneau T, et al. Smoldering multiple myeloma. Blood. 2015;125(20):3069-75. Doi: 10.1182/blood-2014-09-568899
5. Lonial S, Boise LH, Kaufman J, Nooka A, Kaufman J, Muppidi S, et al. How I treat high-risk myeloma. Blood. 2015;126(13):1536-43. Doi: 10.1182/blood-2015-06-653261
6. Oriol A, Motlló C. Nuevos fármacos en el tratamiento del mieloma múltiple. Medicina Clínica. 2014;143(6):268-74. Doi: 10.1016/j.medcli.2013.10.012
7. García Quetglas E, Azanza Perea JR, Lecumberri Villamediana R. Nuevas estrategias terapéuticas en el tratamiento del mieloma múltiple. Análisis de su eficacia y coste-efectividad. Med Clin (Barc). 2008;130(16):626-35. Doi: 10.1157/13120342
8. De Castro R, Carnot J. Mieloma múltiple. Diagnóstico, estudio y tratamiento. Manual prácticas médicas - Hospital Hermanos Ameijeiras [internet]. 2013 [citado 2018 jun 14]. Disponible en: https://www.sld.cu/galerias/pdf/sitios/hematologia/mmarreglado.pdf
9. Corre J, Munshi N, Avet-Loiseau H, Landgren O, Kyle R, Pfeiffer R, et al. Genetics of multiple myeloma: another heterogeneity level? Blood. 2015;125(12):1870-6. Doi: 10.1182/blood-2014-10-567370
10. Maiolino A, Magalhães RJP. Mieloma múltiplo e insuficiência renal. Rev Bras Hematol Hemoter. 2007;29(1):86-91. Doi: 10.1590/S1516-84842007000100016
Notas de autor
* Autor de correspondencia: renanramirezv@hotmail.com