
Revista Ciencias de la Salud
ISSN:1692-7273 | eISSN:2145-4507
ISSN:1692-7273 | eISSN:2145-4507

Co-Expression Network Analysis Identifies Possible Hub Genes in Aging of the Human Prefrontal Cortex
Análisis de redes de coexpresión identifica posibles genes clave en el envejecimiento de la corteza prefrontal humana
Análise de redes de co-expressão identifica possíveis genes chave no envelhecimento do córtex pré-frontal humano
César Payán-Gómez, Julián Riaño-Moreno, Diana Amador-Muñoz, Sandra Ramírez-Clavijo
Co-Expression Network Analysis Identifies Possible Hub Genes in Aging of the Human Prefrontal Cortex
Revista Ciencias de la Salud, vol. 17, no. 2, 2019
Universidad del Rosario
Julián Riaño-Moreno 1
El Bosque University, Colombia
Diana Amador-Muñoz
Universidad del Rosario, Colombia
Sandra Ramírez-Clavijo
Universidad del Rosario, Colombia
Received: 04 December 2018
Accepted: 15 March 2019
Additional information
To cite this article: Payán-Gómez C, Riaño-Moreno J, Amador-Muñoz D, Ramírez-Clavijo S. Co-expression Network Analysis Identifies Possible Hub Genes in Aging of the Human Prefrontal Cortex. Rev Cienc Salud. 2019;17(2):201-22. Doi: https://doi.org/10.12804/10.12804/revistas.urosario.edu.co/revsalud/a.7924
Abstract: Introduction: Aging is the main risk factor for the development of chronic diseases such as cancer, diabetes, Parkinson’s disease, and Alzheimer’s disease. The central nervous system is particularly susceptible to progressive functional deterioration associated with age, among the brain regions the prefrontal cortex (pfc) has one of the highest involvements. Transcriptomics studies of this brain region have identified the decrease in synaptic function and activation of neuroglia cells as fundamental characteristics of the aging process. The aim of this study was to identify hub genes in the transcriptomic deregulation in the pfc aging to advance in the knowledge of this process. Materials and methods: A gene co-expression analysis was carried out for 45 people 60 to 80 years old compared with 38 people 20 to 40 years old. The networks were visualized and analyzed using Cytoscape; citoHubba was used to determine which genes had the best topological characteristics in the co-expression networks. Results: Five genes with high topological characteristics were identified. Four of them —hpca, cacng3, ca10, plppr4— were repressed and one was over-expressed —cryab—. Conclusion: The four repressed genes are expressed preferentially in neurons and regulate the synaptic function and the neuronal plasticity, while the overexpressed gene is typical of glial cells and is expressed as a response to neuronal damage, facilitating myelination and neuronal regeneration.
Keywords Aging, prefrontal cortex, transcriptomics, gene coexpression networks, wgcna, hub genes.
Resumen: Introducción: el envejecimiento es el principal factor de riesgo para el desarrollo de enfermedades crónicas como el cáncer, la diabetes, el Parkinson y el Alzheimer. El sistema nervioso central es particularmente susceptible al deterioro funcional progresivo asociado con la edad, entre las regiones cerebrales con mayor compromiso se encuentra la corteza prefrontal (cpf). Estudios de transcriptómica de esta región han identificado como características fundamentales del proceso de envejecimiento la disminución de la función sináptica y la activación de las células de la neuroglia. No es claro cuáles son las causas iniciales, ni los mecanismos moleculares subyacentes a estas alteraciones. El objetivo de este estudio fue identificar genes clave en la desregulación transcriptómica en el envejecimiento de la cpf para avanzar en el conocimiento de este proceso. Materiales y métodos: se hizo un análisis de coexpresión de genes de los transcriptomas de 45 personas entre 60 y 80 años con el de 38 personas entre 20 y 40 años. Las redes fueron visualizadas y analizadas usando Cytoscape, se usó citoHubba para determinar qué genes tenían las mejores características topológicas en las redes de coexpresión. Resultados: se identificaron cinco genes con características topológicas altas. Cuatro de ellos —hpca, cacng3, ca10, plppr4— reprimidos y uno sobreexpresado —cryab—. Conclusión: los cuatro genes reprimidos se expresan preferencialmente en neuronas y regulan la función sináptica y la plasticidad neuronal, mientras el gen sobreexpresado es típico de células de la glía y se expresa como respuesta a daño neuronal facilitando la mielinización y la regeneración neuronal.
Palabras clave: envejecimiento, corteza prefrontal, transcriptómica, redes de coexpresión de genes, wgcna, genes clave.
Resumo: Introdução: o envelhecimento é o principal fator de risco pra o desenvolvimento de doenças crónicas como o câncer, a diabetes, o Parkinson e o Alzheimer. O sistema nervoso central é particularmente susceptível ao deterioro funcional progressivo associado à idade, uma das regiões do cérebro com maior compromisso é o pré-frontal (cpf). Estudos de transcritoma desta região têm identificado como características fundamentais do processo de envelhecimento a diminuição da função sináptica e ativação das células da neuroglia. Não é claro quais são as causas iniciais, nem os mecanismos moleculares subjacentes a estas alterações. O objetivo deste estudo foi identificar genes chave na desregulação transcritoma no envelhecimento da cpf para avançar no conhecimento deste processo. Materiais e métodos: se fez uma análise de co-expressão de genes dos transcritomas de 45 pessoas entre 60 e 80 anos com o de 38 pessoas entre 20 e 40 anos. As redes foram visualizadas e analisadas usando Cytoscape, usou-se citoHubba para determinar que genes tinham as melhores características topológicas nas redes de co-expressão. Resultados: identificaram-se cinco genes com características topológicas altas. Quatro deles —hpca, cacng3, ca10, plppr4— reprimidos e um superexpresso —cryab—. Conclusão: os quatro genes reprimidos se expressam preferencialmente em neurônios e regulam a função sináptica e plasticidade neuronal, enquanto o gene superexpresso é típico de células da glia e se expressa como resposta ao dano neuronal facilitado a mielinização e a regeneração neuronal.
Palavras-chave: envelhecimento, córtex pré-frontal, transcritoma, redes de co-expressão de genes, wgcna, genes chave.
Introduction
Aging is a complex phenotype that develops through the intervention of various cellular and molecular processes (1). Some of these processes are related to the reaction to the damage accumulation in the dna, senescent cells number increase, mitochondrial dysfunction, stem cells depletion, epigenetic modifications, alterations in the cellular communication, and proteostasis loss (1). One of the tissues most vulnerable to the effects of aging is the central nervous system (cns).
The cns is particularly susceptible to the decline in the function over the years, since its recovery is limited by the presence of post-mitotic cells. Structural and functional modifications associated with aging have been described in the cns. Images obtained through magnetic resonance evidence a decrease of the gray matter (2). Histologic analyses report, beside dendritic regression and signs of neuronal death, especially in the prefrontal cortex (pfc), also demyelination of the white substance, and dna oxidative damage in the oligodendrocytes (3). In general, neurons undergo cell death due to energy deprivation, mitochondrial dysfunction, calcium overload and the impact of disproportionate inflammatory processes (4). With advancing age, dna oxidative damage and increase of its activation state in glia cells have been identified (5). All this leads to a decline of the cns functions and to an increase in the vulnerability to suffer neurodegenerative diseases (1). Not all brain regions age in the same way; it has been observed that the pfc is more susceptible to exhibit characteristics associated with aging, for this reason, it has been subject of various researches in order to identify which processes are altered by aging in this zone (6).
With aging, the speed of the information processing and of the capacity of short-term and long-term memory decreases; besides, there is an alteration in the impulse control, all these functions depend as much on the modification of the impulse control, as on the integrity of the pfc, the hippocampus and the post-rolandic cortex too (7, 8, 9, 10, 11). Transcriptomics studies of aged pfc have consistently reported decrease of the synapsis function and activation of the neuroglia cells (12,13,14,15). Recently, modifications in the splicing mechanisms associated with the transcriptome in the pfc have been found (16). However, it is not clear which are the initial causes or the molecular mechanisms underlying these alterations.
One step forward in understanding the molecular processes involved in aging is the identification of the hub genes in the transcriptional deregulation associated to the process. To accomplish this, a weighted correlation network analysis (wgcna) was conducted. This is a data mining procedure used in analyzing biological networks based on paired correlations between variables (17). For this specific research, wgcna was used to identify central genes in the modification of the aging transcriptome, thus comparing the transcriptomes of 45 persons aged between 60 and 80 years and the transcriptomes of 38 persons aged between 20 and 40 years. Five genes with high topologic characteristics were identified. Four of them —hpca, cacng3, ca10, plppr4— were repressed and one overexpressed cryab. The four repressed genes are expressed preferentially in neurons and regulate the synaptic function and the neuronal plasticity, while the overexpressed gene is characteristic for neuroglia cells, its expression is activated as a response to neuronal damage and facilitates myelinization and regeneration. Identifying these hub genes underlines the role of the synaptic dysfunction in brain aging and offers potential biomarkers and therapeutic targets for modifying the normal and pathological aging processes.
Materials and Methods
Data Selection: A search in the transcriptomics database geo omnibus (https://www.ncbi.nlm.nih.gov/geo/) on researches studying the expression of prefrontal cortex genes in humans throughout life and with a high number of biological replies. For this purpose, key words were used such as human prefrontal cortex, aging, lifespan. Selection criteria for obtaining a data set were original data availability (raw data with no previous processing), and that the study had at least 30 biological replies in each interest age group: 20 to 40 years old and 60 to 80 years old. Samples should be taken from healthy people, with no decreased mental status nor neurological diseases at the moment of death.
Quality Control: For each microarray a percentage of probesets with fluorescence, background fluorescence, rna degradation profiles, intensities density histogram; and establishment of standard errors failures were identified. Microarrays with negative outcomes in at least one of the measured parameters were not included in the subsequent analysis. Quality control was conducted through the qc.affy function of the sympleaffy package used in R (https://www.r-project.org/) (18).
Correction of the Batch Effect: In studies in which a large number of samples is analyzed and, furthermore, samples are difficult to collect, as in the case of human brain tissue, it is common that samples are processed in different days. Affymetrix microarrays are very sensitive to variations of the conditions under which hybridization is conducted, therefore, this possible confusion factor should always be considered. Identifying the batch effect was carried out by recovering the hybridization date of each microarray through the pdata command of the affydata package implemented in R program for statistical analysis (https://www.r-project.org/) (19). Once the different microarray hybridization dates (batches) have been identified, this non-biological variability source was corrected by using a methodology based on nonparametric Bayesian statistics implemented in ComBat (20). The extent of the batch effect correction was assessed by a main component analysis (mca). mca is a technique for dimensionality decline that allows identifying unbiasedly the natural relation between the samples composed of different variables. Thus, it is expected that, if the effect of a confusion factor (the same batch) produces important modifications in the transcriptome, the samples that share the same batch in a natural way should be closer to each other than even to other that share the same experimental group, in the case of this research, age.
Identifying Differentially Expressed Genes: Microarrays were assigned to two different experimental groups: elderly (aged 60-80 years) and young (aged 20-40 years). Besides, all were simultaneously normalized using rma. rma algorithm was also used for determining the expression levels of each gene in the microarray. In order to detect differential expression genes (deg) in the two experimental groups, the limma statistics package (lineal models for microarray data) was used (21). With this methodology the expression change was calculated and the statistical significance adjusted for multiple comparisons (fdr - false discovery rate). It was a priori established that a gene is differentially expressed if an expression change is higher than |1.2| and a fdr value lower than 0.05.
Biological Interpretation of the Genes: An overrepresentation analysis of the biological pathways was performed using the david tool with the database of biological process pathways of gene ontology (go) (22). Repressed and overexpressed ged were analyzed independently in order to have an intuitive interpretation of the deregulation direction of the altered pathway.
Gene Coexpression Networks Analysis ( wgcna ): Coexpression networks were developed based on the ged between older and young. The similarity matrix was calculated, by identifying the Pearson correlation coefficients of the expression levels along all samples for all possible gene pairs. The similarity threshold was then identified, it was calculated with the adjacency function that is established according to the unique characteristics of each similarity matrix (23). In order to select the threshold, the adaptable method developed by Elo was used (24). The method compares the grouping coefficient of the network (Co) with the grouping coefficient expected for a random network (Cr) by different tao values. Finally, the adjacency matrix of the network was established, this is a 2x2 matrix that allows the representation of binary relationships, in this case it represents if a pair of genes do coexpress (1) or do not (0). All processes for the wgcna were performed in an R environment (https://www.r-project.org/) using statistical functions embedded in the same environment.
Visualizing and Analyzing Coexpression Networks: For the visualization of the coexpression networks Cytoscape open code program was used, and also for ulterior studies on this matter different algorithms implemented in the same program have been used (25). The topological characteristics of all genes were established according to ten different algorithms: six of them are global descriptors (bottleneck, closeness, grouping coefficient, eccentricity, radiality, stress), and the remaining four are local descriptors (intercalation, degree, dmnc, mnc) (26). A gene was identified in the network as being a hub if it was among the first ten genes with the highest values for at least seven of the ten studied topologic characteristics. It was decided that seven measures will be the cutting point, since six are global descriptors, therefore, if a gene is among the ten best measures of at least seven descriptors, this gene is important within the network, globally as well as locally.
Validation of Hub Genes for Coexpression Networks: Genes identified as hub genes in the coexpression networks were validated against the results of a meta-analysis of four independent transcriptomics studies on human pfc in which the transcriptomes of persons between 60 and 80 years old with persons between 20 and 40 years old were compared for deg identification (15).
Results
Quality Control and Differential Expression Genes Identification in Elderly and Young People
The dataset with the highest number of biological replies within the ages of interest (60 to 80 years old and 20 to 40 years old) in geo omnibus was gse71620 (10). In this study, the expression levels of all pcf genes, Brodmann 11 (ba11) were analyzed. All persons included in the study were neurologically healthy at the time when the brain tissue was collected. Measuring the gene expression level was performed with the Affimetrix “Human Gene 1.1 st Array” microarray. After quality control, four microarrays were removed (three of the group of elderly and one of the group of young) because they had expression levels very different to the remaining data. It was also identified that the dataset had been hybridized in 19 different dates, this potential variation source was adjusted with ComBat (20). Figure 2 shows the non-biased distribution of the samples under study in this research: 45 samples of the group of elderly and 38 samples of the group of young.

Figure 1 shows the two principal components (pc) containing the greatest variation of the dataset, as a whole they represent 18 % of variability.
pc1 represents the largest part of the separation of the samples of the group of the elderly contrasted with the group of the young. Although there is no categorical separation between groups, the sample distribution shows that there are differences in the gene expression in the pfc with regard to age. Young tend to be located towards the positive side of the Pc1, while elderly towards the negative side of the mc1. Samples mixed with the opposite group also show the variability in individual aging processes and that there are also differences between chronological and biological age.
When comparing transcriptomes of the elderly with those of the young, 276 repressed genes and 166 overexpressed genes were identified, amounting to a total of 442 deg out of the 18 840 genes in the microarray. With these genes, with different expressions between the study groups, the subsequent analyses were conducted.
In order to establish the biological relevance of the deg, a pathway overrepresentation analysis was performed using david, repressed genes and overexpressed genes were analyzed separately, thus making it possible to identify the direction of change of the altered biological pathway (27). Table 1 represents the ten pathways with more significant p values. In the group of the repressed genes it was found that some genes took part in biological processes characteristic of the neurons, such as synaptic transmission, ion transport, glutamate signaling pathway, and some biological processes related to conduct and perception, such as sensory pain perception and memory (table 1A). In the group of overexpressed genes, pathways related to the development of astrocytes, myelinization and inflammation were found as being overrepresented (table 1B).
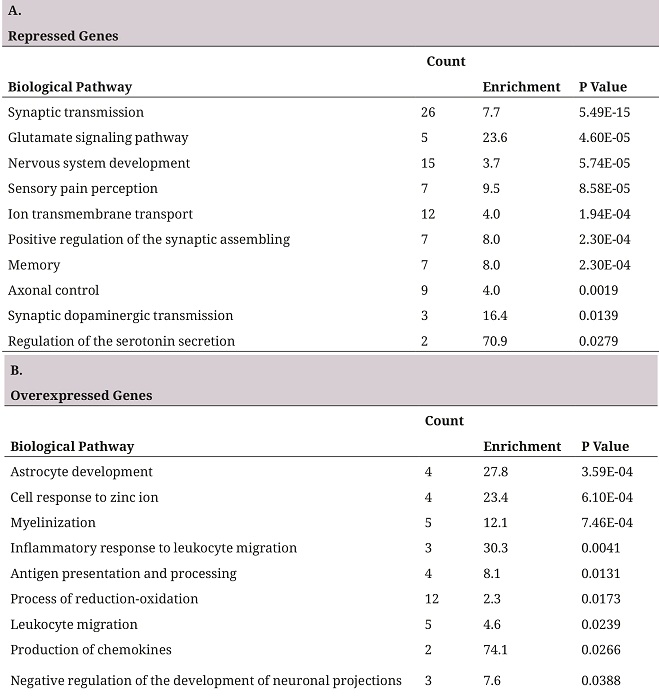
A. First ten pathways overrepresented in the list of repressed genes. B. First ten pathways overrepresented in the list of overexpressed genes. The biological pathways correspond to the database of go biological processes. The Counting column shows the deg number in the pathway, Enrichment indicates how many times there are more genes than the randomly expected.
Development of the Weighted Coexpression Networks
With the expression levels of the 442 deg in the 83 samples a correlation matrix was developed, then the similarity threshold was identified and the adjacency matrix was computed. The similarity threshold was calculated through the maximum local method, and it was found that a Pearson correlation coefficient equal or higher than 0.75 differentiates the distributions of the correlations between the study genes and those of a random gene population (23). This threshold allowed to separate genes with low correlation coefficients from genes with the highest correlation coefficient, 251 genes out of 442 deg were selected.
With the 251 highly correlated genes, an adjacency matrix was developed and, on this base, the coexpression networks were visualized in Cytoscape (25). Figure 2 shows the coexpression networks for the deg between elderly and young. Three networks were produced, the largest one (figure 2A) is composed of 158 repressed genes. In the second largest network, composed of 89 overexpressed genes (figure 2B), two subnetworks are identified: one that is highly connected and composed of 68 genes, and another smaller network, with a lower connection level, composed by 21 genes. Additionally, there is a network of four overexpressed genes connected to one another (figure 2C). Five deg did not connect to any of the networks described before.
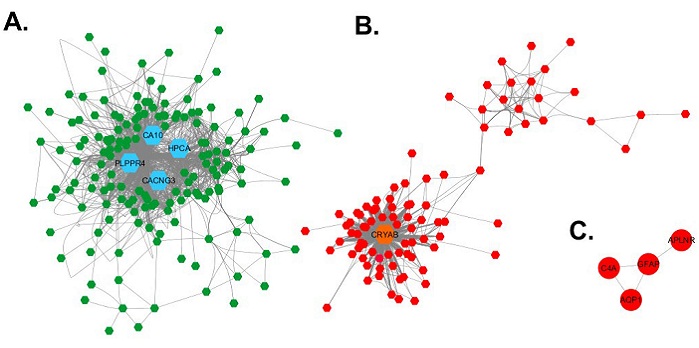
Visualization of the coexpression networ in Cytoscape. A. Coexpression network of repressed genes. B. Coexpression network of overexpressed genes. C. Network of four overexpressed genes. The nodes are represented as hexagons, each node correspond to one gene, the lines connecting a pair of nodes represent a Pearson correlation coefficient higher than 0.75. The repressed genes are represented in green and the overexpressed in red. In each network the hub genes are represented for their high topological level by bigger hexagons, colored in blue the repressed genes, and in orange the overexpressed genes.
In order to identify the hub genes in the coexpression networks, a topological characterization of these by cytoHubba was performed (26). The two networks with the highest number of genes (figure 2A and table 2A) were topologically characterized by ten different algorithms (table 2), the genes with higher outcomes for at least seven of the ten measures are selected as hub genes. Four repressed genes in the coexpression network were identified (figure 2A and table 2A), and one in the overexpressed genes network (figure 2B and table 2B).
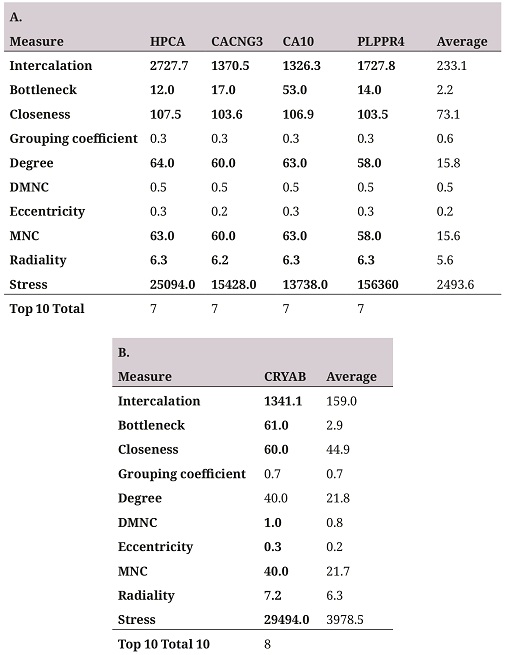
A. Hub genes selected in the repressed gene coexpression network. B. Hub genes selected in the overexpressed gene coexpression network. The Average column shows the average of the measure for all genes in the network as reference point. The Top 10 Total column represents the number of measures in which the selected gene was among the ten highest scores for the ten topological measures.
As an external validation of the results, the expression change direction of the hub genes was compared with the results of a meta-analysis in which four independent studies different to the one used in the present research were integrated, these characterized the transcriptomic response to aging in the pfc (15). Of the five identified hub genes, three were analyzed in the meta-analysis —ca10, plppr4, cryab— and all of them presented the same direction of change found in this study, the first two repressed, and the third one, overexpressed. These results suggest that the identified genes are reliable and that the result could be generalized to the aging process of the pfc in humans. The remaining two genes that were not studied in the meta-analysis — hpca and cacng3— had the best values in the topological measures in the coexpression network of repressed genes. Due to the high consistency to the genes that were studied in the meta-analysis, the latter were conserved although it was not possible to confirm their direction of change with the external study.
Finally, based on the detected values in the microarrays for each hub gene in all the studied samples, a two-ways heatmap was developed (figure 3); this in order to determine the capacity of the hub genes to differentiate older from young and to know the distribution of the expression levels in each analyzed person.
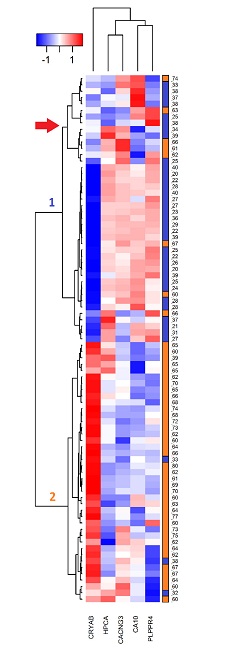
The heatmap was developed by clustering genes by similarity in the expression levels through the samples (vertical clustering), and grouping the samples by similarity in the gene expression levels (horizontal clustering). The group of the elderly is represented in orange and the group of the young in blue; the numbers of the right vertical bar correspond to the ages of the people in the study. The intensity level of each gene was normalized with a Z transformation (left superior bar); the blue color represents the low expression level, while the red color represents the high expression.
In the heatmap the repressed genes were organized in a cluster, the only overexpressed gene (cryab) remained outside this cluster. This gene has the most homogeneous expression levels among the samples of the same experimental group.
Samples were organized in two clusters, cluster 1 was mainly composed of the samples of the young persons (34 of this experimental group), and seven of elderly. This cluster, in turn, is organized in two clusters, a larger one where the distribution of the hub gene intensities is homogeneous, and another smaller cluster, marked with a red arrow in Figure 3, composed of nine samples of young and five of elderly. In that cluster, although the behavior of the hub genes is similar to almost all samples of the young, in one or two genes by sample the expression change direction is different to almost all samples of its age group. Cluster 2 is predominantly composed of 38 samples of the group of elderly and of three samples of the group of young.
If cluster 1 is of the samples of the young individuals and cluster 2 of the samples of the elderly, in sum there are eleven samples in the opposite group, these correspond to 13.3 % of the analyzed persons. This way, combining the expression of the five hub genes allows the correct classification of 86.7 % of people, according to the experimental group.
Discussion
Aging appears in heterogeneous ways in humans, since it is associated with genetic, environmental and random factors that cannot be totally controlled in this kind of researches (14, 28,29,30). In pca (figure 1) this heterogeneity was evidenced since, when unbiasedly identifying the natural relations within the transcriptome in samples of persons of the two groups at the opposite ends of life expectancy, it was found that, although a high percentage of young persons can clearly be differentiated from the older, it is not possible to totally separate both experimental groups. There is a transition zone shared by young and elderly, even further, some young people mix with older persons and vice versa. It is tempting to propose that the persons in the group of the young who are nearer to the negative pc1 zone (figure 1) are people whose biological age is higher than the chronological age, while the persons of the group of the elderly located in positive pc1 regions were in the opposite situation: a biological age lower than their effective chronological age. Unfortunately, the experimental design of our study does not allow the verification of this hypothesis since the samples were obtained from post mortem brains and it is therefore not possible to track each one of them.
When the transcriptomes of the group of the elderly and those of the younger were contrasted, 442 deg were found. When taking age as a dichotomic variable (older or young) the identified genes represent the transcriptional deregulation common to most persons analyzed between ages 60 and 80 years. In order to identify the central processes secondary to these modifications, the coexpression networks analysis was performed. 251 (56 %) of the 442 deg had a coexpression level higher than the level to be expected randomly, hence, almost half of the deg presented expression patterns with low correlation to other deg, this, again, can be explained by the heterogeneity of aging and by the stochastic nature of some processes associated with it, such as the accumulation of dna mutations over the years that would produce expression patterns not consistent among different samples, for the pfc of each person will have its own random mutation accumulation history (31, 32).
Of the 251 coexpressed deg, 246 genes were included in three coexpression networks, indicating that this gene group has a similar behavior along the analyzed samples, and that they could represent a common transcriptional response in pfc aging. All repressed genes were located in only one network (figure 2A) while most overexpressed genes were located in two connected networks and in a network composed of four genes (figures 2B and 2C).
The four genes network (figure 2C) was composed of the genes that codify for the glial fibrillary acid protein (gfap), aquaporin 1 (aqp1), the acidic form of the complement factor 4 (c4a), and the apelin receptor (aplnr). The first two genes are recognized markers of the activation of astrocytes in response to nervous tissue damage. For instance, gfap is overexpressed in astrocytes in response to trauma or ischemia and in neurodegenerative diseases, additionally, an increase in its expression during aging has been consistently reported since the 90’s (33, 34). Something similar occurs with the aqp1 gene that is expressed during the astrocyte polarization in response to brain damage, and in neurodegenerative diseases such as Parkinson’s (35,36,37). Another gene of the network, C4a, is part of the humoral immune response; it has been proved that microglia cells express it during the formation of amyloid plaques in Alzheimer’s disease (38). Besides, there is a clear increase in its expression associated with a higher age (39). Regarding the last gene in the network, aplnr, it has been recently reported that it acts as an important maintenance regulator mediated by the endothelium of glioblastoma derived cells; and in the same sense, in mice it was found that aplnr is crucial for astrocyte maturation through endothelial cell signaling action (40,41,42). In the overrepresented pathways in the group of overexpressed aging genes, astrocyte development and inflammatory pathways (table 1B) were found, both are connected to the four genes of this network. Three of the four genes take direct part on astrocyte activation, one of the best known and defined processes of brain aging (12, 43). This indicates that the approach used in the present study is useful for finding relevant processes for the phenotype of interest and can identify relationships between molecular processes that interact as pfc aging determinants.
In the two coexpression networks with the highest number of genes (figure 2A and Figure 2B) a search for hub genes was conducted. The rationality behind identifying this type of genes is that it is probable that, having high topological characteristics, these genes control important points in the modulation of a coexpression network and, therefore, they will be important for the studied biological process (44, 45). In the repressed genes network (figure 4A) four hub genes were identified: hypocalcin (hpca), calcium voltage-gated channel auxiliary subunit gamma 3 (cacng3), carbonic anhydrase-related protein 10 (ca10), and phospholipid phosphatase related 4 protein (plppr4). Hypocalcin is part of the family of specific neuron proteins that bind to calcium, their function is related to the regulation of the voltage dependent calcium channels (46). Another protein found in the network was cacng3, which is specifically expressed in the brain, it is assumed that its function is to stabilize inactive calcium channels (47). It has been described that polymorphisms in this gene are associated with events of absence seizures in childhood (48). In the hippocampus of rats it was found that with aging there is a decrease of the regulating protein expression levels of the neurotransmission networks, among them hpca and cacng3 (49). Our outcomes underline the importance of the decline of the synaptic function with aging and the central role of the calcium regulation in the transcriptomic modifications of the pfc over time. In the overrepresented pathways of the repressed genes group, synaptic transmission, signaling of glutamate receptor and memory were identified (table 1A), all these pathways in relation to the processes mediated by hpca and cacng3m proteins.
The third hub gene of the coexpression network of repressed genes is ca10, this gene codifies for a protein that catalyzes the reversible hydration of carbon dioxide in various biological processes, among them the development of the brain (50). ca10 protein binds directly to neurexin 1 (nrxn1) and promotes the expression in the pre-synaptic membrane surface of the α and β neurexins; this process is relevant for the establishment and specification of synaptic connections (51). Interestingly, we did not find a significative decrease of the nrxn1 expression levels, ca10 primary ligand, this could indicate that the pathway alteration depends on the decline of ca10 signaling and not on an upstream alteration of it, a fact that stresses the central and regulating role of ca10 in this neuronal aging mechanism.
The last repressed hub gene is plppr4, it is part of the phospholipid phosphatases whose function is to catalyze dephosphorylation of bioactive lipidic mediators regulating different cell functions (52). plppr4 is expressed in neurons and is found specifically in growing axons. It has been proved that it plays an important role during the development and formation of new axonal terminals, the formation of filopodia, the extension of neurites, and the axonal reorganization after neural damage (53, 54).
Our analysis identified the repression of two hub genes related to the neuronal function, specifically to establishing new synapsis (ca10), and the neuronal regeneration (plppr4). The fact that they are repressed in the pfc of individuals older than 60 years old explains, at least partially, the reasons why the neuronal dysfunction and the loss of cognitive functions appear. This, additionally, is supported on the fact that one repressed pathway of the deg analysis was the development of the nervous system (table 1A).
In the overexpressed gene network (figure 2B) only one hub gene was identified: Alpha-crystallin B chain (cryab). cryab is part of the family of small heat shock proteins that act as molecular chaperones. These chaperones bind to misfolded proteins to prevent their aggregation, they can also inhibit apoptosis and contribute to intracellular architecture (55). cryab is expressed both in astrocytes and in oligodendrocytes and it has been found that the overexpression of this gene in astrocytes decreases the neurological deterioration in mouse models of Huntington disease through the decrease of the aggregation of mutant huntingtin protein (56). Also in an animal model of glaucoma, a degenerative disease of the optical nerve, a neuroprotective effect with CRYAB injection in the vitreous was observed. However, cryab has been also connected to the development of cns pathology, since it has been reported that in substantia nigra of persons with Parkinson’s it is overexpressed, indicating a possible participation in the dopaminergic neuronal degeneration (57, 58). Likewise, it has been informed that cryab increases astrocyte mediated demyelination in multiple sclerosis and that it increases oxidation of the beta amyloid protein, thus incrementing its aggregating potential and therefore its neurotoxicity (59, 60). Unlike the former result, with the induction of the cryab expression in astrocytes, the beta amyloid protein aggregation is inhibited (61). These contradictory results, where cryab acts sometimes as a neuroprotective protein and in other cases as neurotoxic could be explained by the post-translational regulation of the protein. cryb can be phosphorylated in different amino acids; phosphorylation of 19, 45 and 59 serines does not confer citoprotection, while phosphorylation of 59 and 45 serines does (59, 62,63,64). In the altered pathways in the overexpressed genes, astrocyte development and myelinization were found de/regulated, although we couldn’t identify the status of cryab phosphorylation due to the experimental design of our study, the fact that the myelinization pathway is increased and that, as previously described, cryab is involved in favoring this process in the peripheral nervous system, could suggest that the increase of the cryab expression in the pfc exerts a cytoprotective effect (65, 66). However, additional studies are needed to define the protein real function in normal aging.
In conclusion, in the present study, five hub genes were identified in the transcriptomic deregulation secondary to pfc aging. Four of these five genes dominate the coexpression network of repressed genes and are involved mainly in the synaptic function and the neuronal plasticity. The only overexpressed hub gene could be exerting a neuroprotective function in response to the synaptic dysfunction.
With the implemented methodological design, it was intended that identifying the genes that presented differential expression when comparing the group of young with the group of elder would allow describing common cell and molecular events in the studied samples. The fact that, when grouping the samples according to the expression levels of the five hub genes, a good discrimination between older and young (figure 3) was allowed, indicates that the selected hub genes could be candidates to biomarkers of biological processes generalizable for pfc aging. These results need to be validated in animal models and other human cohorts with normal and pathological aging. Once they are validated, they could be used as a basis for evaluating potential biomarkers of biological age, as prevention and treatment targets of neurodegenerative diseases, and for evaluating anti-aging therapies.
Contribution of the Authors
Study Conception: César Payán-Gómez, Diana Amador-Muñoz and Sandra Ramírez-Clavijo. Data Analysis: César Payán-Gómez. Research: Julián Riaño-Moreno. Methodology: César Payán-Gómez. Results Analysis and Document Drafting: César Payán-Gómez, Julián Riaño-Moreno, Diana Amador-Muñoz, Sandra Ramírez-Clavijo.
Disclaimer
The authors declare that there is no conflict of interests.
References
1.López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153(6):1194-217. Doi: 10.1016/j.cell.2013.05.039
2.Dumitriu D, Hao J, Hara Y, Kaufmann J, Janssen WG, Lou W, et al. Selective changes in thin spine density and morphology in monkey prefrontal cortex correlate with aging-related cognitive impairment. J Neurosci. 2010;30(22):7507-15. Doi: 10.1523/JNEUROSCI.6410-09.2010
3.Hawrylycz MJ, Lein ES, Guillozet-Bongaarts AL, Shen EH, Ng L, Miller JA, et al. An anatomically comprehensive atlas of the adult human brain transcriptome. Nature. 2012;489(7416):391-9. Doi: 10.1038/nature11405
4.Primiani CT, Ryan VH, Rao JS, Cam MC, Ahn K, Modi HR, et al. Coordinated gene expression of neuroinflammatory and cell signaling markers in dorsolateral prefrontal cortex during human brain development and aging. PLoS One. 2014;9(10):e110972. Doi: 10.1371/journal.pone.0110972
5.Cornejo F, von Bernhardi R. Age-dependent changes in the activation and regulation of microglia. Adv Exp Med Biol. 2016;949:205-26. Doi: 10.1007/978-3-319-40764-7_10
6.Tisserand DJ, Pruessner JC, Sanz Arigita EJ, van Boxtel MP, Evans AC, Jolles J, et al. Regional frontal cortical volumes decrease differentially in aging: an MRI study to compare volumetric approaches and voxel-based morphometry. Neuroimage. 2002;17(2):657-69. Doi: https://doi.org/10.1006/nimg.2002.1173
7.Woodcock EA, Greenwald MK, Khatib D, Diwadkar VA, Stanley JA. Pharmacological stress impairs working memory performance and attenuates dorsolateral prefrontal cortex glutamate modulation. Neuroimage. 2018. Doi: 10.1016/j.neuroimage.2018.11.017
8.Lemaitre H, Goldman AL, Sambataro F, Verchinski BA, Meyer-Lindenberg A, Weinberger DR, et al. Normal age-related brain morphometric changes: nonuniformity across cortical thickness, surface area and gray matter volume? Neurobiol Aging. 2012;33(3):617.e1-9. Doi: 10.1016/j.neurobiolaging.2010.07.013
9.Boros BD, Greathouse KM, Gearing M, Herskowitz JH. Dendritic spine remodeling accompanies Alzheimer’s disease pathology and genetic susceptibility in cognitively normal aging. Neurobiol Aging. 2019;73:92-103. Doi: 10.1016/j.neurobiolaging.2018.09.003
10.Chen CY, Logan RW, Ma T, Lewis DA, Tseng GC, Sibille E, et al. Effects of aging on circadian patterns of gene expression in the human prefrontal cortex. Proc Natl Acad Sci U S A. 2016;113(1):206-11. Doi: 10.1073/pnas.1508249112
11.Chung HK, Tymula A, Glimcher P. The Reduction of Ventrolateral Prefrontal Cortex Gray Matter Volume Correlates with Loss of Economic Rationality in Aging. J Neurosci. 2017;37(49):12068-77. Doi: 10.1523/JNEUROSCI.1171-17.2017
12.Clarke LE, Liddelow SA, Chakraborty C, Münch AE, Heiman M, Barres BA. Normal aging induces A1-like astrocyte reactivity. Proc Natl Acad Sci U S A. 2018;115(8):E1896-E905. Doi: 10.1073/pnas.1800165115
13.Dillman AA, Majounie E, Ding J, Gibbs JR, Hernandez D, Arepalli S, et al. Transcriptomic profiling of the human brain reveals that altered synaptic gene expression is associated with chronological aging. Sci Rep. 2017;7(1):16890. Doi: 10.1038/s41598-017-17322-0
14.Harris SE, Riggio V, Evenden L, Gilchrist T, McCafferty S, Murphy L, et al. Age-related gene expression changes, and transcriptome wide association study of physical and cognitive aging traits, in the Lothian Birth Cohort 1936. Aging (Albany NY). 2017;9(12):2489-503. Doi: 10.18632/aging.101333
15.Payán-Gómez C, Rodríguez D, Amador D, Ramírez S. Integrative analysis of global gene expression 2 identifies opposite patterns of reactive astrogliosis in 3 aged human prefrontal cortex 4. aging. 2018;1:33. Doi: 10.20944/preprints201810.0733.v1
16.Raj T, Li YI, Wong G, Humphrey J, Wang M, Ramdhani S, et al. Integrative transcriptome analyses of the aging brain implicate altered splicing in Alzheimer’s disease susceptibility. Nat Genet. 2018;50(11):1584-92. Doi: 10.1038/s41588-018-0238-1
17.Zhao W, Langfelder P, Fuller T, Dong J, Li A, Hovarth S. Weighted gene coexpression network analysis: state of the art. J Biopharm Stat. 2010;20(2):281-300. Doi: 10.1080/10543400903572753
18.Miller C. Simpleaffy: Very simple high level analysis of Affymetrix data; 2018.
19.Gautier L. Affydata: Affymetrix Data for Demonstration Purpose. R package version 1.30.0. 2018.
20.Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8(1):118-27. Doi: 10.1093/biostatistics/kxj037
21.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47. Doi: 10.1093/nar/gkv007
22.Huang dW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44-57. Doi: 10.1038/nprot.2008.211
23.Zhang B, Horvath S. A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol. 2005;4:Article17. Doi: 10.2202/1544-6115.1128
24.Elo LL, Järvenpää H, Oresic M, Lahesmaa R, Aittokallio T. Systematic construction of gene coexpression networks with applications to human T helper cell differentiation process. Bioinformatics. 2007;23(16):2096-103. Doi: 10.1093/bioinformatics/btm309
25.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498-504. Doi: 10.1101/gr.1239303
26.Chin CH, Chen SH, Wu HH, Ho CW, Ko MT, Lin CY. cytoHubba: identifying hub objects and sub-networks from complex interactome. BMC Syst Biol. 2014;8 Suppl 4:S11. Doi: 10.1186/1752-0509-8-S4-S11
27.Maere S, Heymans K, Kuiper M. BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics. 2005;21(16):3448-9. Doi: 10.1093/bioinformatics/bti551
28.Unnikrishnan A, Freeman WM, Jackson J, Wren JD, Porter H, Richardson A. The role of dna methylation in epigenetics of aging. Pharmacol Ther. 2018. Doi: 10.1016/j.pharmthera.2018.11.001
29.Gladyshev VN. Aging: progressive decline in fitness due to the rising deleteriome adjusted by genetic, environmental, and stochastic processes. Aging Cell. 2016;15(4):594-602. Doi: 10.1111/acel.12480
30.Taylor AM, Pattie A, Deary IJ. Cohort Profile Update: The Lothian birth cohorts of 1921 and 1936. Int J Epidemiol. 2018;47(4):1042-r. Doi: 10.1093/ije/dyy022
31.Burla R, La Torre M, Merigliano C, Vernì F, Saggio I. Genomic instability and dna replication defects in progeroid syndromes. Nucleus. 2018;9(1):368-79. Doi: 10.1080/19491034.2018.1476793
32.Frenk S, Houseley J. Gene expression hallmarks of cellular ageing. Biogerontology. 2018;19(6):547-66. Doi: 10.1007/s10522-018-9750-z
33.Hol EM, Pekny M. Glial fibrillary acidic protein (gfap) and the astrocyte intermediate filament system in diseases of the central nervous system. Curr Opin Cell Biol. 2015;32:121-30. Doi: 10.1016/j.ceb.2015.02.004
34.Kohama SG, Goss JR, Finch CE, McNeill TH. Increases of glial fibrillary acidic protein in the aging female mouse brain. Neurobiol Aging. 1995;16(1):59-67.
35.Badaut J, Brunet JF, Grollimund L, Hamou MF, Magistretti PJ, Villemure JG, et al. Aquaporin 1 and aquaporin 4 expression in human brain after subarachnoid hemorrhage and in peritumoral tissue. Acta Neurochir Suppl. 2003;86:495-8.
36.Badaut J, Fukuda AM, Jullienne A, Petry KG. Aquaporin and brain diseases. Biochim Biophys Acta. 2014;1840(5):1554-65. Doi: 10.1016/j.bbagen.2013.10.032
37.Hoshi A, Tsunoda A, Tada M, Nishizawa M, Ugawa Y, Kakita A. Expression of aquaporin 1 and aquaporin 4 in the temporal neocortex of patients with Parkinson’s disease. Brain Pathol. 2017;27(2):160-8. Doi: 10.1111/bpa.12369
38.Emmerling MR, Watson MD, Raby CA, Spiegel K. The role of complement in Alzheimer’s disease pathology. Biochim Biophys Acta. 2000;1502(1):158-71. Doi: https://doi.org/10.1016/S0925-4439(00)00042-9
39.Zhou J, Fonseca MI, Pisalyaput K, Tenner AJ. Complement C3 and C4 expression in C1q sufficient and deficient mouse models of Alzheimer’s disease. J Neurochem. 2008;106(5):2080-92. Doi: 10.1111/j.1471-4159.2008.05558.x
40.Harford-Wright E, Gavard J. Apelin, the devil inside brain tumors. J Exp Neurosci. 2018;12:1179069518759680. Doi: 10.1177/1179069518759680
41.Harford-Wright E, Andre-Gregoire G, Jacobs KA, Treps L, Le Gonidec S, Leclair HM, et al. Pharmacological targeting of apelin impairs glioblastoma growth. Brain. 2017;140(11):2939-54. Doi: 10.1093/brain/awx253
42.Sakimoto S, Kidoya H, Naito H, Kamei M, Sakaguchi H, Goda N, et al. A role for endothelial cells in promoting the maturation of astrocytes through the apelin/APJ system in mice. Development. 2012;139(7):1327-35. Doi: 10.1242/dev.072330
43.Palmer AL, Ousman SS. Astrocytes and aging. Front Aging Neurosci. 2018;10:337. Doi: 10.3389/fnagi.2018.00337
44.Miller JA, Horvath S, Geschwind DH. Divergence of human and mouse brain transcriptome highlights Alzheimer disease pathways. Proc Natl Acad Sci U S A. 2010;107(28):12698-703. Doi: 10.1073/pnas.0914257107
45.Miller JA, Woltjer RL, Goodenbour JM, Horvath S, Geschwind DH. Genes and pathways underlying regional and cell type changes in Alzheimer’s disease. Genome Med. 2013;5(5):48. Doi: 10.1186/gm452
46.Helassa N, Antonyuk SV, Lian LY, Haynes LP, Burgoyne RD. Biophysical and functional characterization of hippocalcin mutants responsible for human dystonia. Hum Mol Genet. 2017;26(13):2426-35. Doi: 10.1093/hmg/ddx133
47.Moss FJ, Dolphin AC, Clare JJ. Human neuronal stargazin-like proteins, gamma2, gamma3 and gamma4; an investigation of their specific localization in human brain and their influence on CaV2.1 voltage-dependent calcium channels expressed in Xenopus oocytes. BMC Neurosci. 2003;4:23. Doi: 10.1186/1471-2202-4-23
48.Everett KV, Chioza B, Aicardi J, Aschauer H, Brouwer O, Callenbach P, et al. Linkage and association analysis of CACNG3 in childhood absence epilepsy. Eur J Hum Genet. 2007;15(4):463-72. Doi: 10.1038/sj.ejhg.5201783
49.VanGuilder HD, Yan H, Farley JA, Sonntag WE, Freeman WM. Aging alters the expression of neurotransmission-regulating proteins in the hippocampal synaptoproteome. J Neurochem. 2010;113(6):1577-88. Doi: 10.1111/j.1471-4159.2010.06719.x
50.Pastorekova S, Parkkila S, Pastorek J, Supuran CT. Carbonic anhydrases: current state of the art, therapeutic applications and future prospects. J Enzyme Inhib Med Chem. 2004;19(3):199-229. Doi: 10.1080/14756360410001689540
51.Sterky FH, Trotter JH, Lee SJ, Recktenwald CV, Du X, Zhou B, et al. Carbonic anhydrase-related protein CA10 is an evolutionarily conserved pan-neurexin ligand. Proc Natl Acad Sci U S A. 2017;114(7):E1253-E62. Doi: 10.1073/pnas.1621321114
52.Sciorra VA, Morris AJ. Roles for lipid phosphate phosphatases in regulation of cellular signaling. Biochim Biophys Acta. 2002;1582(1-3):45-51. Doi: https://doi.org/10.1016/S1388-1981(02)00136-1
53.Bräuer AU, Savaskan NE, Kühn H, Prehn S, Ninnemann O, Nitsch R. A new phospholipid phosphatase, PRG-1, is involved in axon growth and regenerative sprouting. Nat Neurosci. 2003;6(6):572-8. Doi: 10.1038/nn1052
54.Trimbuch T, Beed P, Vogt J, Schuchmann S, Maier N, Kintscher M, et al. Synaptic PRG-1 modulates excitatory transmission via lipid phosphate-mediated signaling. Cell. 2009;138(6):1222-35. Doi: 10.1016/j.cell.2009.06.050
55.Yamamoto S, Yamashita A, Arakaki N, Nemoto H, Yamazaki T. Prevention of aberrant protein aggregation by anchoring the molecular chaperone αB-crystallin to the endoplasmic reticulum. Biochem Biophys Res Commun. 2014;455(3-4):241-5. Doi: 10.1016/j.bbrc.2014.10.151
56.Oliveira AO, Osmand A, Outeiro TF, Muchowski PJ, Finkbeiner S. αB-Crystallin overexpression in astrocytes modulates the phenotype of the BACHD mouse model of Huntington’s disease. Hum Mol Genet. 2016;25(9):1677-89. Doi: 10.1093/hmg/ddw028
57.Anders F, Liu A, Mann C, Teister J, Lauzi J, Thanos S, et al. The small heat shock protein α-crystallin B shows neuroprotective properties in a glaucoma animal model. Int J Mol Sci. 2017;18(11). Doi: 10.3390/ijms18112418
58.Liu Y, Zhou Q, Tang M, Fu N, Shao W, Zhang S, et al. Upregulation of alphaB-crystallin expression in the substantia nigra of patients with Parkinson’s disease. Neurobiol Aging. 2015;36(4):1686-91. Doi: 10.1016/j.neurobiolaging.2015.01.015
59.Kuipers HF, Yoon J, van Horssen J, Han MH, Bollyky PL, Palmer TD, et al. Phosphorylation of αB-crystallin supports reactive astrogliosis in demyelination. Proc Natl Acad Sci U S A. 2017;114(9):E1745-E54. Doi: 10.1073/pnas.1621314114
60.Narayanan S, Kamps B, Boelens WC, Reif B. alphaB-crystallin competes with Alzheimer’s disease beta-amyloid peptide for peptide-peptide interactions and induces oxidation of Abeta-Met35. FEBS Lett. 2006;580(25):5941-6. Doi: 10.1016/j.febslet.2006.09.063
61.Ren Z, Yang M, Guan Z, Yu W. Astrocytic α7 nicotinic receptor activation inhibits amyloid-β aggregation by upregulating endogenous αB-crystallin through the PI3K/Akt signaling pathway. Curr Alzheimer Res. 2018. Doi: 10.2174/1567205015666181022093359
62.Li R, Reiser G. Phosphorylation of Ser45 and Ser59 of αB-crystallin and p38/extracellular regulated kinase activity determine αB-crystallin-mediated protection of rat brain astrocytes from C2-ceramide- and staurosporine-induced cell death. J Neurochem. 2011;118(3):354-64. Doi: 10.1111/j.1471-4159.2011.07317.x
63.Morrison LE, Hoover HE, Thuerauf DJ, Glembotski CC. Mimicking phosphorylation of alphaB-crystallin on serine-59 is necessary and sufficient to provide maximal protection of cardiac myocytes from apoptosis. Circ Res. 2003;92(2):203-11.
64.Chis R, Sharma P, Bousette N, Miyake T, Wilson A, Backx PH, et al. α-crystallin B prevents apoptosis after H2O2 exposure in mouse neonatal cardiomyocytes. Am J Physiol Heart Circ Physiol. 2012;303(8):H967-78. Doi: 10.1152/ajpheart.00040.2012
65.Zigmond RE. Heat shock protein that facilitates myelination of regenerating axons. Proc Natl Acad Sci U S A. 2017;114(9):2103-5. Doi: 10.1073/pnas.1700755114
66.Lim EF, Nakanishi ST, Hoghooghi V, Eaton SE, Palmer AL, Frederick A, et al. AlphaB-crystallin regulates remyelination after peripheral nerve injury. Proc Natl Acad Sci U S A. 2017;114(9):E1707-E16. Doi: 10.1073/pnas.1612136114
Author notes
1
Faculty of Medicine, Cooperative University of
Colombia, Villavicencio, Colombia. Department of Bioetics, El Bosque University,
Bogotá, Colombia
* Corresponding author: cesar.payan@urosario.edu.co




