Revista Ciencias de la Salud
ISSN:1692-7273 | eISSN:2145-4507
ISSN:1692-7273 | eISSN:2145-4507

Medicamentos biosimilares en Colombia: una revisión desde el consumo informado
Biosimilar Medicines in Colombia: An Approach from the Informed Consumption
Medicamentos biossimilares na Colômbia: uma revisão desde o consumo informado
Diana Rocío Bernal-Camargo, Julio César Gaitán-Bohórquez, Édgar Iván León-Robayo
Medicamentos biosimilares en Colombia: una revisión desde el consumo informado
Revista Ciencias de la Salud, vol. 16, núm. 2, 2018
Universidad del Rosario
Diana Rocío Bernal-Camargo * diana.bernalc@urosario.edu.co
Universidad del Rosario, Colombia
Julio César Gaitán-Bohórquez **
Universidad del Rosario, Colombia
Édgar Iván León-Robayo ***
Universidad del Rosario, Colombia
Recibido: 12 Mayo 2017
Aceptado: 02 Febrero 2018
Información adicional
Para citar este artículo: Bernal-Camargo DR, Gaitán-Bohórquez
JC, León Robayo ÉI. Medicamentos biosimilares en Colombia: una revisión desde
el consumo informado. Rev Cienc Salud. 2018;16(2):311-339.
Doi: https://doi.org/10.12804/revistas.urosario.edu.co/revsalud/a.6772
Resumen: Introducción: este artículo presenta un análisis sobre medicamentos biosimilares en Colombia con miras a establecer si existe un consumo informado respecto de estos fármacos, teniendo en cuenta la calidad y cantidad de información que circula en relación con su disponibilidad y acceso económico (precios). Desarrollo: el tipo de estudio es cualitativo con un diseño documental a partir de la revisión en bases de datos como Pubmed, Scopus, Scielo, Vlex y Redalyc, bases de información de registros sanitarios de agencias reguladoras de medicamentos (FDA, EMA e Invima) y el Sistema de Información de Vademecum Med Informática. Uno de los biosimilares autorizados en FDA y cuatro de la EMA se encuentran registrados en el Invima; sin embargo, son más los biosimilares no autorizados ni en FDA ni en EMA que se encuentran registrados en el Invima, los cuales aún no aparecen con esta clasificación de biosimilares ni en la agencia reguladora colombiana ni en ningún otro medio oficial o comercial. De los medicamentos biológicos de referencia de estos biosimilares, 12 tienen control de precios y 7 han sido autorizados al menos una vez vía judicial a los usuarios del sistema. Conclusiones: si bien, la reglamentación ha sido de gran avance en relación con el acceso a biosimilares y, en consecuencia, a sus biológicos pioneros, el sistema aún tiene barreras jurídicas, de información, disponibilidad y acceso que dificultan la protección y efectividad del derecho a la salud de la población en términos de un consumo informado de estos.
Palabras clave: medicamentos biosimilares, medicamentos biológicos, consumo informado, derecho a la salud, propiedad intelectual, agencias reguladoras de medicamentos, barreras de acceso a medicamentos.
Abstract: Introduction: This article presents an analysis of biosimilar drugs in Colombia, aiming at critically analyzing whether the consumption of biosimilar drugs in Colombia is fully informed or if it rather is characterized for its lack of information. Informed consumption in the sense of taking into account the quality and quantity of the information circulating in relation to the availability and affordability (prices) of biosimilar drugs. Development: This is a qualitative documentary analysis, based on the review of databases such as Pubmed, Scopus, Scielo, Vlex and Redalyc, and sanitary databases of drug´s regulatory agencies (FDA, EMA and the Colombian Invima) and the Vademecum Med Informatica. One of the biosimilars authorized by FDA and four of those by EMA were also registered before by the Invima. However, the number of Invima authorized biosimilars is higher than that authorized by FDA and EMA. It is also important to highlight the fact that any biosimilar is not registered as such neither before the Invima nor before any other official or commercial source. Out of the biological reference products with biosimilars, twelve have regulated price and seven have been authorized at least once via court ruling. Conclusions: Although the regulations are progressive in relation to the access to biosimilars and, consequently, to their biological pioneers, the system still has legal, availability and information barriers that undermine the protection and effectiveness of the right to health.
Keywords: biosimilar drugs, biological medicines, informed consumption, the right to health, intellectual property rights, drug regulatory agencies, barriers to access to medicines.
Resumo: Introdução: este artigo apresenta uma análise sobre medicamentos biossimilares na Colômbia com vista a estabelecer se existe um consumo informado respeito destes fármacos, tendo em conta a qualidade e quantidade de informação que circula em relação com a sua disponibilidade e acesso econômico (preços). Desenvolvimento: o tipo de estudo é qualitativo com um desenho documental a partir da revisão em bases de dados como Pubmed, Scopus, Scielo, Vlex e Redalyc, bases de informação de registros sanitários de agências reguladoras de medicamentos (FDA, EMA e Invima) e o Sistema de Informação de Vademecum Med Informática. Um dos biossimilares autorizados em FDA e 4 da EMA, se encontram registrados no Invima, no entanto são mais os biossimilares não autorizados nem na FDA nem na EMA que se encontram registrados no Invima, os quais ainda não aparecem com esta classificação de biossimilares nem na agência reguladora colombiana, nem em nenhum outro meio oficial ou comercial. Dos medicamentos biológicos de referência destes biossimilares, 12 têm controle de preços e 7 têm sido autorizados pelo menos uma vez via judicial aos usuários do sistema. Conclusões: se bem a regulamentação tem sido de grande avanço em relação com o acesso a biossimilares e, consequentemente, a seus biológicos pioneiros, o sistema ainda tem barreiras jurídicas, de informação, disponibilidade e acesso que dificultam a proteção e efetividade do direito à saúde da população em termos de um consumo informado dos mesmos.
Palavras-chave: medicamentos biossimilares, medicamentos biológicos, consumo informado, direito à saúde, propriedade intelectual, agências reguladoras de medicamentos, barreiras de acesso a medicamentos.
Introducción
La biotecnología aplicada a la generación de medicamentos ha permitido desarrollar medicamentos biológicos, obtenidos a partir de material de origen biológico. Una vez expira su patente, se abre paso a la generación de medicamentos biosimilares, que son así altamente similares a los biológicos de referencia (1, 2). En el sistema jurídico colombiano, el Ministerio de Salud y Protección Social (MSPS) estableció un sistema regulatorio específico para “el registro sanitario de medicamentos biológicos nuevos (pioneros) y conocidos (biocompetidores, biosimilares o biogenéricos)”, por medio del Decreto 1782 de 2014 (3). La norma estableció tres tipos de rutas para demostrar las condiciones de calidad, seguridad y eficacia, que tienen en común la presentación de una información básica conforme al contenido del artículo 6° del Decreto 1782/14 4 .
Para los medicamentos biológicos nuevos o pioneros se estableció la denominada ruta de expediente completa, que obliga a presentar los estudios preclínicos (in-vivo y/o in-vitro) y los ensayos clínicos, además de que deben cumplir con los requisitos comunes para sus pertinentes autorizaciones. Los medicamentos biológicos conocidos tienen dos rutas: la de comparabilidad y la abreviada de comparabilidad. La primera implica la presentación de resultados de un ejercicio de comparabilidad entre el biológico objeto de evaluación y el biológico de referencia. La segunda, por su parte, permite respaldar la solicitud de registro con la información disponible sobre seguridad y eficacia de un conjunto de medicamentos que contienen un ingrediente farmacéutico activo altamente similar (3, 4).
Estos medicamentos —tanto los biológicos como los biosimilares— se analizan en este artículo, en el que se discute la perspectiva del acceso a medicamentos en el contexto del derecho de toda persona al disfrute del más alto nivel posible de salud física y mental. Este debate se ha observado desde dos perspectivas: desde el enfoque de la protección a la propiedad intelectual, en cabeza de los desarrolladores, y respecto de los derechos humanos, como garantía de protección del derecho a la salud de las personas.
Este análisis, en el fondo, plantea un conflicto de derechos que se da entre la necesidad del sistema de patentes en el sector farmacéutico como mecanismo de compensación a los altos costos de Investigación y Desarrollo (I+D), y el desequilibrio de las condiciones para acceder a los medios —en este caso los medicamentos— que buscan el goce más alto del derecho a la salud (5, 6).
Es decir, los avances en biotecnología para la generación de nuevos medicamentos se traducen en resultados patentables, lo que conlleva un desequilibrio entre los países desarrollados y subdesarrollados, puesto que las potencias económicas poseen el control sobre los mercados biotecnológicos (7). Por ejemplo, algunos informes han señalado que existen aproximadamente “4275 empresas biotecnológicas en el mundo, de ellas: el 64 % trabaja en la salud humana, el 45 % están en Norteamérica, el 38 % en Europa y el 17 % en Asia y el Pacífico” (8). Como se ha mencionado antes, el desarrollo de medicamentos biosimilares se da gracias a la expiración de la patente de los medicamentos biológicos pioneros, obtenida a partir de procesos de I+D protegidos por el sistema de patentes.
Algunos resultados de investigaciones publicadas en la materia dan cuenta de la incidencia del acceso a medicamentos en relación con el derecho a la salud, en los que se destaca cómo la regulación del Estado incide positivamente en el acceso a estos y cómo las vías judiciales se constituyen en los mecanismos alternos de acceso (9). Por ejemplo, acorde con un análisis realizado sobre la judicialización del derecho al acceso a medicamentos en el caso colombiano para el periodo 2011-2012, casi el 20 % de las acciones de tutela se referían a este derecho (10).
En esta medida, se evidencia cómo el derecho al acceso a medicamentos implica las garantías en sí mismas de accesibilidad y disponibilidad, en donde el Estado puede tomar medidas para efectivizarlo, como lo muestra Dorado-Torres en el estudio de caso sobre Ecuador, en el que, mediante el Decreto Ejecutivo 118 de 2009 y la Resolución 10-04-9-iepi de 2010, concedió cinco licencias obligatorias para medicamentos de alto costo (11). En sentido similar, se encuentra el caso brasileño, donde la potestad de esa flexibilidad fue utilizada en el 2007 para el Efavirenz, o el caso colombiano, en que el MSPS, mediante la Resolución 2475 del 14 de junio de 2016, hizo la declaratoria de interés público del principio activo Imatinib (Glivec®) con el efecto inmediato de ordenar su regulación de precio, que ha sido un obstáculo para garantizar su acceso a la población (12, 13). La justificación de este tipo de licencias es la de tratar de anteponer el interés público a los derechos exclusivos de ámbito privado que otorga la patente.
Estas posiciones teóricas responden a un choque de dos posturas entre dos grandes organizaciones internacionales involucradas en la materia, esto es, entre la Organización Mundial de la Salud (OMS) y la Organización Mundial del Comercio (OMC). La primera hace un paulatino trabajo relacionado con la conceptualización del acceso a medicamentos como un derecho humano, en contraste con la OMC, que protege los intereses empresariales tomando como punto de partida los Acuerdos sobre los Aspectos de los Derechos de Propiedad Intelectual (Adpic), que conllevan implicaciones importantes en el campo farmacéutico (14).
Tales discusiones en el campo de los biológicos y los biosimilares, como lo ha expuesto Martín Uribe, replantean el papel del Estado, quien se encuentra conminado a propiciar y estimular el mercado de los biosimilares, debate que se agudizó durante su proceso de regulación en el país (15).
En términos de acceso a biosimilares, la protección y promoción del derecho a la salud debe enfocarse hacia el consumo informado de medicamentos (CIM). En tal medida, este artículo tiene como objetivo analizar y evaluar el tipo de la información (calidad y suficiencia) que sobre los biosimilares se encuentra disponible para los usuarios del sistema de salud, por medio de las plataformas oficiales de las instituciones públicas relacionadas con el tema.
En este sentido, se acoge la visión del documento de consenso acordado por el Grupo Europeo del Proyecto de Acceso y Adquisición de Mercado de los Productos Biosimilares, y adoptado por el Grupo Directivo del Proceso sobre Responsabilidad Corporativa en el campo de los productos farmacéuticos. El instrumento considera de gran importancia las “cuestiones relativas a la mejora de la información disponible sobre el concepto de biosimilares”, como asuntos importantes para la toma de decisiones por parte de profesionales de salud, sociedades científicas, autoridades nacionales y locales competentes; y, por supuesto, los pacientes y las asociaciones de pacientes. Si bien, estos no tienen en todos los casos posibilidades de elección respecto de los medicamentos biológicos y/o biosimilares disponibles, sí tienen el derecho a acceder a toda la información posible respecto de estos por medio de las autoridades sanitarias (16).
De esta manera, se pretende aportar al debate sobre biológicos y biosimilares, elementos relacionados con el derecho a la información —como garantía del derecho al acceso a los medicamentos—, para lo cual se identifica el estado actual de disponibilidad, en términos de información, de estos medicamentos en el contexto colombiano.
Este enfoque hacia el CIM es pertinente, puesto que se ha descrito que uno de los factores del uso inapropiado de medicamentos está asociado con el uso de fármacos innecesariamente caros, en la medida que
En muchos países la gente escoge los medicamentos por su marca comercial. Los productos de marcas registradas con frecuencia son más caros que el medicamento genérico que contiene el mismo producto. Además, las personas no saben que dos medicamentos con distinto nombre comercial pueden contener exactamente la misma sustancia. El precio de los medicamentos es un elemento importante para los consumidores (17).
Por esta razón, es importante evaluar la calidad de la información disponible sobre este tipo de medicamentos, lo cual permite tomar decisiones informadas a los consumidores e, incluso, a los profesionales de la salud y, por su puesto, a los dispensadores de medicamentos.
Para tal fin, en primer lugar se hace una aproximación teórica y reglamentaria de medicamentos biológicos y biosimilares mediante la literatura académica y las normas jurídicas de las agencias reguladoras objeto de revisión, y de la normativa colombiana, así como una aproximación al sistema de patentes y protección de medicamentos, como quiera que cuando estas expiran se genera la oportunidad para la generación de los biosimilares. En segundo término, se presenta y analiza el estado actual de la información disponible sobre los medicamentos biosimilares en Colombia, a partir del contraste de la información de las bases de registro de biosimilares aprobados por la FDA (Food and Drug Administration) y por EMA (European Medicines Agency) que aparecen en el Invima (Instituto Nacional de Vigilancia de Medicamentos y Alimentos), y se hace con esto una relación de cada medicamento sobre su estado de registro, indicaciones, control de precios (en casos de existir) y acceso vía mecanismo judicial (acción de tutela con pronunciamiento de la Corte Constitucional o la Corte Suprema de Justicia), para establecer si esta información es accesible a los usuarios o si la que se encuentra disponible es suficiente y adecuada para garantizar el derecho al CIM.
El tipo de estudio que se desarrolló en esta investigación fue de tipo cualitativo con un diseño documental, a partir de tres fases: la primera, de construcción teórica, que permitió hacer una revisión de la literatura científica existente sobre medicamentos biológicos y medicamentos biosimilares, relacionada con su definición y normativa aplicable. Para la búsqueda se emplearon las bases de datos Pubmed, Scopus, Scielo, Vlex y Redalyc y se incluyeron los términos MeSH: medicamentos biosimilares, medicamentos biológicos, acceso a medicamentos, regulación de precios, propiedad intelectual y regulaciones. Las bases de datos electrónicas consultadas fueron PubMed, Scielo y EBSCO. La segunda fase consistió en el rastreo de los medicamentos biosimilares disponibles en Estados Unidos (EE. UU.), la Unión Europea (UE) y Colombia (COL). La búsqueda se hizo para el caso de EE. UU., por medio del sistema de información de la FDA; para la UE, por medio del sistema de información de la EMA; y para el caso COL, por medio del sistema de información de Invima, termómetro de precios del MSPS, Sismed, Datos Abiertos Colombia y Vademecum Med Informática. La tercera fase consistió en identificar la información disponible en Colombia para cada uno de los biosimilares encontrados y determinar su calidad y suficiencia para dar respuesta a la pregunta sobre si esta información garantizaba el derecho al CIM de biológicos y biosimilares en el país.
Desarrollo
Medicamentos biológicos y biosimilares
Los medicamentos biológicos son fármacos sintetizados a partir de moléculas complejas, producidos en organismos vivos, que resultan de difícil replicación exacta (1). Estos medicamentos han tomado mayor auge en el tratamiento de enfermedades graves, lo que genera una serie de esperanzas para su tratamiento.
Tanto en la literatura científica como en la normativa de diferentes países no existe consenso en la distinción entre medicamentos biotecnológicos y los biológicos. Quienes lo hacen, como es el caso de la Organización Panamericana de la Salud, entienden a los primeros como una categoría dentro de los biológicos, que se obtienen a través de tecnología de ADN recombinante (18). Los segundos, por su parte, son definidos como aquellas medicinas que se obtienen a partir de microorganismos, sangre y otros tejidos vivos, cuya producción puede incluir: “uso de células eucariotas, sustancias biológicas extraídas de tejidos, incluidos los tejidos humanos, animales y vegetales, productos obtenidos por tecnología de ADN recombinante o hibridoma, propagación de microorganismos en embriones o animales, entre otros” (18).
Por su parte, los medicamentos biosimilares surgen con ocasión de la expiración de las patentes de los biológicos y, pese a las diferentes definiciones que se han aportado, se pueden entender como aquellos productos altamente similares a los de referencia (biológicos), con algunas diferencias en relación con los componentes clínicamente inactivos, sin diferencias clínicas significativas en relación con la seguridad, la pureza y la potencia, entre el biosimilar y el biológico (2).
Las diferentes regulaciones y agencias reguladoras de medicamentos tienen sus propias definiciones, que pueden estar en consonancia o tener una relativa diferencia con las conceptualizaciones dadas por la comunidad académica, y mencionadas en líneas previas.
Acorde con la sección 351 del Public Health Service Act, la FDA, entiende por medicamentos biológicos los:
virus, suero terapéutico, toxina, antitoxina, vacuna, sangre, componente sanguíneo o derivado, producto alergénico, proteína (excepto cualquier polipéptido sintetizado químicamente), o producto análogo, o arsfenamina o derivado de arsfenamina (o cualquier otro arsénico orgánico trivalente) —pound, aplicable a la prevención, tratamiento o curación de una enfermedad o condición del ser humano (19, 20, 21).
En la misma normativa, la FDA entiende a los biosimilares como aquellos:
productos biológicos altamente similares a un producto biológico de referencia específico, a pesar de las diferencias menores en compuestos clínicamente inactivos, de modo que no existen diferencias clínicamente significativas entre el producto biológico de referencia y el producto biológico que ha sido autorizado como biosimilar (19, 20, 21).
Lo anterior, en términos de seguridad, pureza y potencia del producto, como se evidencia en la sección 42 U.S.C. § 262 (k), de esta norma general. En el sistema jurídico estadounidense se distinguen con la expresión: follow-on biologics (22).
Por su parte, la EMA se fundamenta en lo dispuesto por la Directiva 2001/83/ce, que define un medicamento biológico como:
[…] un producto cuyo principio activo es biológico. Una sustancia biológica es aquella que se produce o se extrae a partir de una fuente biológica y que necesita, para su caracterización y determinación de su calidad, una combinación de ensayos físico-químico y biológicos junto con el proceso de producción y su control. Se considerarán medicamentos biológicos: los medicamentos inmunológicos y los medicamentos derivados de la sangre o el plasma humanos, tal y como se definen en los apartados 4 y 10 del artículo 1; los medicamentos que pertenezcan al ámbito de aplicación de la parte A del anexo del Reglamento (CEE) No 2309/93; los medicamentos de terapia avanzada, definidos en la parte iv de este anexo (23).
En relación con los medicamentos biosimilares, la directiva no trae una definición en estricto sentido; sin embargo, la EMA en uno de sus documentos guía los define así:
Un medicamento biológico similar, conocido como ‘biosimilar’, es un producto similar a un medicamento biológico que ya ha sido autorizado llamado “medicamento de referencia”. El principio activo de un medicamento biosimilar es un principio activo biológico conocido, y similar a la del medicamento de referencia. A un producto biológico similar y a su correspondiente medicamento de referencia se les presupone el mismo perfil de eficacia y seguridad, y son generalmente utilizados como tratamiento en las mismas condiciones (24).
En el caso del sistema colombiano, por medio de su Decreto 1782 de 2014, el MSPS distingue entre medicamentos biológicos, biotecnológicos y biosimilares. En su artículo 3.1., los primeros son definidos como todos aquellos:
Medicamentos derivados de organismos o células vivas o sus partes. Se pueden obtener de fuentes tales como tejidos o células, componentes de la sangre humana o animal (como antitoxinas y otro tipo de anticuerpos, citoquinas, factores de crecimiento, hormonas y factores de coagulación), virus, microorganismos y productos derivados de ellos como las toxinas. Estos productos son obtenidos con métodos que comprenden, pero no se limitan a cultivo de células de origen humano o animal, cultivo y propagación de microorganismos y virus, procesamiento a partir de tejidos o fluidos biológicos humanos o animales, transgénesis, técnicas de Ácido Desoxirribonucleico (ADN) recombinante, y técnicas de hibridoma. Los medicamentos que resultan de estos tres últimos métodos se denominan biotecnológico (4).
Y en las líneas finales de esta disposición se señala que los medicamentos biotecnológicos son aquellos biológicos que resultan específicamente de los métodos de “transgénesis, técnicas de Ácido Desoxirribonucleico (ADN) recombinante, y técnicas de hibridoma” (4).
Siguiendo la normativa colombiana, el decreto mencionado se refiere a medicamentos biológicos nuevos o pioneros, para los que establece la denominada ruta de expediente completo, y los medicamentos biológicos conocidos, biosimilares, biogenéricos o biocompetidores, entendiéndolos como aquellos que contienen un ingrediente activo altamente similar a un biológico pionero. Para tal fin establece las llamadas rutas de comparabilidad y la abreviada de comparabilidad (3, 4).
Siguiendo la normativa colombiana, el decreto mencionado se refiere a medicamentos biológicos nuevos o pioneros, para los que establece la denominada ruta de expediente completo, y los medicamentos biológicos conocidos, biosimilares, biogenéricos o biocompetidores, entendiéndolos como aquellos que contienen un ingrediente activo altamente similar a un biológico pionero. Para tal fin establece las llamadas rutas de comparabilidad y la abreviada de comparabilidad (3, 4).
De la revisión a la información disponible en la FDA, de los 148 medicamentos biológicos aprobados a diciembre de 2017, 9 son biosimilares que se encuentran autorizados en la actualidad.
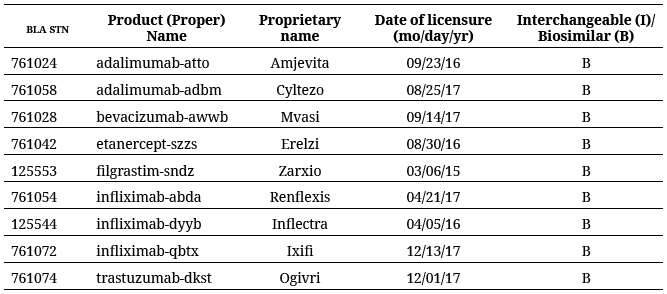
Acorde con la información disponible en la web de la EMA, a la fecha se han presentado 42 solicitudes de medicamentos biosimilares, de las cuales 2 se han rechazado y 3 se han retirado, de forma que 37 se encuentran autorizadas por la Agencia (tabla 2).
En el caso colombiano, como agencia reguladora encargada de otorgar el registro sanitario para estos medicamentos, el Invima es responsable de garantizar la calidad de la información sobre estos medicamentos. Por medio de su página web se puede acceder al listado completo y actualizado de medicamentos que cuentan con registro sanitario. Sin embargo, esta información no clasifica actualmente los medicamentos entre biológicos, biotecnológicos y biosimilares.
Con la entrada en vigencia del Decreto 1782/14 y las correspondientes guías de estabilidad y de evaluación de inmunogenicidad de los medicamentos biológicos se esperaría una mayor información para los usuarios sobre estos productos biológicos, a fin de que esta resulte suficiente y adecuada para su consumo.
El sistema de patentes de productos farmacéuticos
Según relata Rengifo, el sistema de patentes se construyó para proteger en un primer momento las industrias metalmecánica y química. Con posterioridad, la investigación microbiológica y la ingeniería genética se extendieron a otros campos como lo agropecuario y farmacéutico, trayendo consigo nuevos productos y mejorando los existentes (27).
Históricamente, las primeras manifestaciones legales sobre inventos se encuentran en la Venecia del Renacimiento (1474); el estatuto inglés de monopolios de James Stuart (1624), la legislación sobre patentes de Estados Unidos (1790). Esta legislación adquiere carácter internacional en el siglo XIX, con el Convenio de París para la Protección de la Propiedad Industrial (1883) —Ley 178 de 1994—, donde se constituyó un sistema de reivindicación de prioridad 7 de solicitudes presentadas dentro de los 12 meses siguientes a la solicitud inicial (28).
Los medicamentos adquirieron una protección intelectual respecto de sus componentes tan solo a partir de los siglos XVIII y XIX. En esos tiempos, eran llamados ‘nostrum remedium’, perdiendo con el tiempo la primera palabra. A mediados del siglo XIX su fabricación se convirtió en una gran industria, principalmente en EE. UU., al ser elaborados con morfina, opio o cocaína, lo que trajo resultados trágicos y las consecuentes críticas por parte de las asociaciones médicas. Por tal motivo, en 1906 se expidió en ese país una normativa sobre drogas y alimentos, la cual permitió el control gubernamental contra ingredientes no etiquetados o inseguros, publicidad engañosa o prácticas ilegales, como forma de protección y garantía a los derechos a la información y la salud de las personas.
Desde aquel entonces, los medicamentos han sido regulados por los países mediante sus normativas internas, pero en materia de propiedad industrial adquieren una connotación internacional. Por ello, son múltiples los instrumentos que los reglamentan. Así sucede con el Acuerdo de Creación de la OMC, incluyendo el Anexo C, relativo al Adpic (1994) —Ley 170 de 1994—. Este documento constituyó la base teórica del marco jurídico de la Decisión 486 de 2000 de la Comunidad Andina, que constituye la normativa supranacional que rige la materia actualmente para Colombia, Ecuador, Perú y Bolivia (30).
De igual importancia es el Tratado de Cooperación de Washington en materia de Patentes (PCT-1970) —Ley 463 de 1998—, el cual extiende la prioridad del Convenio de París hasta por 31 meses. Sumado a esto, existen normas sobre patentes en los tratados de libre comercio, entre los que se destaca Estados Unidos —Ley 1519 de 2012—, la Asociación Europea de Libre Comercio EFTA —Ley 1372 de 2010— y la Unión Europea —Ley 1669 de 2013—.
Cuando alguien descubre un medicamento capaz de generar alivio a una enfermedad o condición está creando un invento 8 , que supone la intervención del intelecto humano sobre las fuerzas y elementos de la naturaleza, para obtener algún resultado o efecto que “ella misma nunca produciría o, de producirlo, lo haría en condiciones de forma tiempo, y espacio no idénticas a las que derivan de dicha intervención humana” (32, 33). Este proceso es complejo y es claro que el inventor se sirve de un patrimonio científico y cultural preexistente, el cual parte del conocimiento previo de leyes naturales, descubrimientos, prácticas, habilidades, invenciones anteriores, etcétera.
Dada la necesidad de este tipo de creaciones, el Estado ha diseñado el sistema de patentes para fomentar la investigación, construido sobre tres pilares: (i) la promoción de la innovación tecnológica, (ii) la elevación de la competitividad industrial y (iii) el fomento del desarrollo económico.
La colectividad accede inmediatamente al conocimiento de la invención y podrá explotarla con libertad una vez expirado el plazo de protección, que para el caso andino corresponde a 20 años, según dispone el artículo 50 de la Decisión 486. En tal medida, esta información es conocida por el público gracias a la publicación que de esta realiza la oficina de propiedad industrial en los denominados “bancos de patentes” 9 (28, 34).
El inventor obtendrá entonces su patente una vez agote el trámite administrativo correspondiente, cuando su invención cumpla tres requisitos: (i) novedad 10 , (ii) nivel inventivo 11 y (iii) aplicación industrial 12 (29, 35). La doctrina distingue el ‘producto’, consistente en el principio activo de un medicamento (p.a.) y este último es toda sustancia o composición poseedora de propiedades curativas o preventivas de las enfermedades humanas o animales, cuya administración permite establecer un diagnóstico médico o restablecer, corregir o modificar sus funciones orgánicas (36). Es decir, se trata de invenciones patentables cuyo titular, por regla general, es una empresa farmacéutica, aunque esto no excluye a otro tipo de entidades o personas naturales.
Vencida la patente, y al estar disponible al público para su industrialización y comercialización, el empresario podrá proteger el medicamento por medio del régimen de marcas, el cual le brinda reconocimiento a su empresa y producto en el mercado.
Antes de los Adpic, ninguna convención internacional establecía estándares mínimos para la regulación de patentes sobre estos productos. Como consecuencia, “la producción genérica de farmacéuticos protegidos por patentes, las importaciones paralelas y la piratería de drogas patentadas eran bastante comunes”(37). Por ello, su inclusión dentro de la regulación internacional constituyó una victoria muy importante para las compañías.
En desarrollo de tales compromisos internacionales, la legislación colombiana define a los medicamentos en el artículo 2º del Decreto 677 de 1995 como un “preparado farmacéutico obtenido a partir de principios activos, con o sin sustancias auxiliares, presentado bajo forma farmacéutica, que se utiliza para la prevención, alivio, diagnóstico, tratamiento, curación o rehabilitación de la enfermedad”(38). Además, la misma disposición establece que “los envases, rótulos, etiquetas y empaques hacen parte integral del medicamento, por cuanto estos garantizan su calidad, estabilidad y uso adecuado” (38). No obstante, para que sea industrialmente comercializado el medicamento en Colombia se hace preciso obtener una autorización sanitaria por parte del Invima, de conformidad con la normativa vigente (39).
Como se puede observar, resulta de gran interés y debate entender el impacto de la expiración de una patente en el campo de los biológicos, puesto que, en muchos casos, durante la vigencia de la patente, el precio de los medicamentos resulta desproporcionado. Al igual que en el caso de los medicamentos genéricos, para los biosimilares existen, como se anotó en el apartado anterior, regulaciones específicas para su fabricación, aprobación y comercialización, en particular si se toma en cuenta que los biosimilares en estricto sentido no son idénticos al biológico pionero (40).
En relación con las bases y sistemas de información revisados en este estudio, en ninguna de ellas existe información relacionada con el tipo de protección a los derechos de propiedad intelectual de los medicamentos biosimilares y sus biológicos de referencia estudiados, de forma que esta clase de información no es accesible a los consumidores de medicamentos.
Los medicamentos biológicos en Colombia
La regulación general de medicamentos biológicos en Colombia parte de la Política Farmacéutica Nacional, del eje sobre “estímulo a la competencia expresada en la obligatoriedad de la prescripción y las transacciones de medicamentos utilizando siempre su Denominación Común Internacional o nombre científico”, desarrollado en la tercera y séptima línea estratégica, que corresponde a la “revisión, depuración y ajuste de las normas para conseguir el marco regulatorio que se necesita para la implantación de la política” y el “aprovechamiento que la gran diversidad de recursos de nuestra biodiversidad ofrece para promover la investigación en productos biológicos, naturales, biotecnológicos, homeopáticos y alternativos”, respectivamente (41).
Con posterioridad el MSPS expidió el Decreto 1782 de 2014, por medio del cual se definieron “los requisitos para el registro sanitario de medicamentos biológicos nuevos (pioneros) y conocidos (biocompetidores, biosimilares o biogenéricos)”, estableciendo tres maneras de presentar la información para la autorización de ingreso al mercado: “una ruta de expediente completo, para los medicamentos biológicos nuevos y dos rutas para los biológicos conocidos: la ruta de comparabilidad y la ruta abreviada de la comparabilidad, incluyendo, en todos los casos, ensayos propios de inmunogenicidad”, de forma que esa ruta abreviada permite “usar la información disponible sobre seguridad y eficacia de un producto sin necesidad de exponer a animales y humanos a experimentos innecesarios” (3, 4).
Con el objeto de determinar la calidad de la información disponible sobre biosimilares para los usuarios del sistema de salud por medio de las plataformas oficiales de las instituciones públicas (Invima y MSPS) relacionadas con el tema, se hizo la búsqueda en sus sistemas públicos de información. Lo primero que debe evidenciarse es que en ninguna de ellas existe una clasificación que dé cuenta de las categorías de medicamentos entre biológicos y biosimilares, ni siquiera entre fármacos de síntesis química y biosimilares.
Conforme a este primer resultado, debió buscarse manualmente la información, por lo que se tomó el listado de los biosimilares aprobados por la FDA y la EMA y se contrastó con la lista general de los medicamentos aprobados por el Invima, tomando como criterio de búsqueda el principio activo del medicamento 13 .
Adicionalmente, se incorporó el listado de biosimilares fabricados en la India, China, Corea y países de Latinoamérica, entre otros, y aprobados por su agencia reguladora, como quiera que se trata de un mercado que tiene un desarrollo significativo en estos países. Si bien, los biosimilares no han sido aprobados por la agencia de EE. UU. o la europea están entrando con un alto impacto al mercado de medicamentos en Latinoamérica (42).
En esta medida, se encontraron los siguientes biológicos, con determinados biosimilares, aunque no tienen como tal esta denominación:

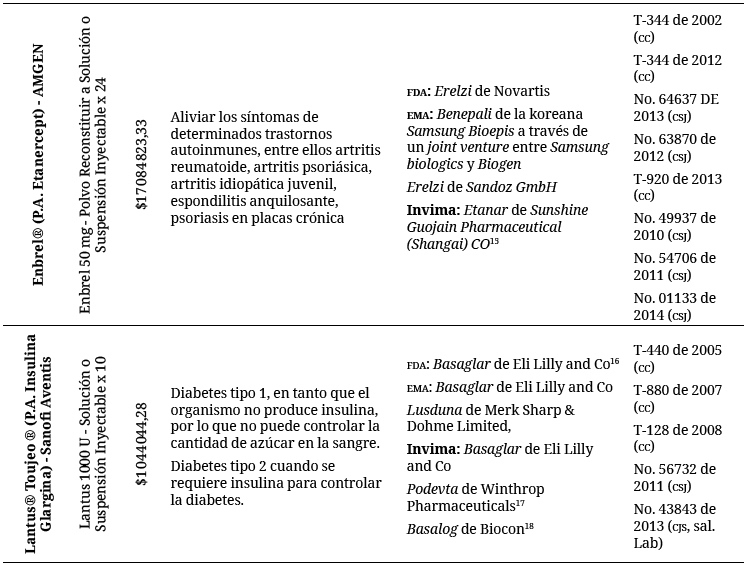





Barreras de acceso a medicamentos biológicos y consumo informado: un análisis de la información encontrada
Conforme a los datos que reposan en los sistemas de información sobre medicamentos en Colombia es importante señalar que esta no es detallada, clara, completa y compresible respecto de medicamentos biológicos y biosimilares. Si bien, es cierto que el MSPS tiene una guía práctica sobre estos, es insuficiente respecto de la clasificación de los que en la actualidad se encuentran autorizados por el Invima en el país. En relación con la información sobre los precios, si bien, existe, se encuentra en otro tipo de base de datos (SISMED) y tampoco clasifica los medicamentos ni da detalles sobre ellos.
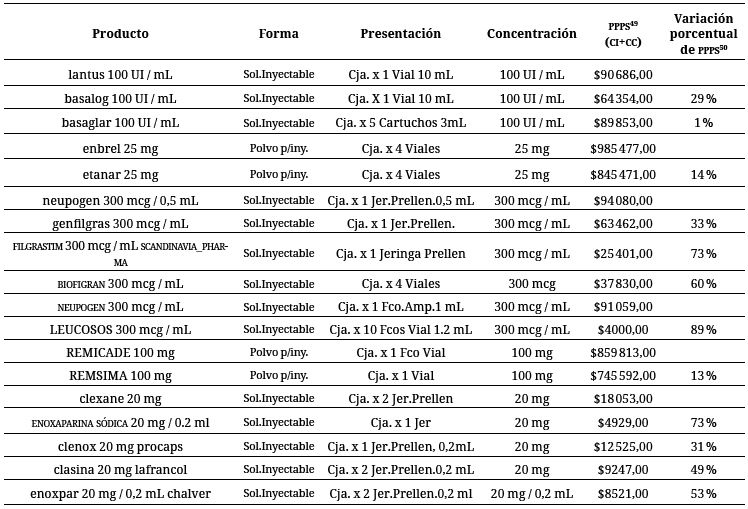
Es de mencionar que tampoco se tiene claridad en relación con la promoción de la industria de biosimilares en el país, en donde actualmente no tienen ninguna participación. En cierta forma, esta situación ha motivado el surgimiento de monopolios en la industria, en donde pueden encontrarse distorsiones económicas relacionadas con el manejo de la oferta y la demanda que dan lugar a conductas como la dosificación de la producción, lo que conlleva al aumento injustificado de precios 51 y que provocaron, precisamente, la intervención del Estado. De los datos reportados en de la tabla 4, en la que se compararon medicamentos biológicos pioneros con algunos de sus biosmilares, a partir de presentaciones del medicamento cuyas dosis son equivalentes, se evidencia que las diferencias de precios entre los biológicos pioneros y los biosimilares pueden ser significativas en términos porcentuales, llegando a una diferencia superior al 50 %, como en el caso Clexane ® y Neupogen ® con sus biosimilares Enoxaparina sódica y Enoxpar, lo cual puede impactar la toma de decisiones dentro del sistema, en especial en el profesional de la salud y el paciente (consumidor), quien no recibe una información clara al respecto.
Esta falta de claridad en el mercado conlleva, a su vez, a “un ámbito de competencia y de rivalidad entre las empresas polarizado en torno a las actividades de investigación e innovación tecnológica” (44).
De ahí la necesidad de nivelar estas ventajas y desventajas en materia de medicamentos al establecer excepciones a los derechos de exclusividad, como lo son el licenciamiento obligatorio 52 y la denominada “disposición de Bolar” 53 . Esta última les permite a los productores de genéricos iniciar el proceso de aprobación ante las autoridades de salud pública para comercializar versiones del producto patentado antes de la expiración de la patente, lo que evita las demoras propias de este tipo de trámites, y que, en principio, aplica también para los biosimilares (37).
El ostensible vínculo entre medicamentos y salud, cuando aquellos son necesarios para la recuperación de esta o para mantener la vida en condiciones de dignidad necesaria para ser vivida o, en extremo, para conservarla, fundamenta la relevancia constitucional de atender de manera privilegiada aspectos como la investigación, la producción y, centralmente, el acceso. Dada su alta incidencia en la garantía de los derechos humanos, cuyo disfrute depende del acceso a los medicamentos en condiciones económicamente razonables, hace necesaria la intervención del Estado mediante la regulación de los precios, la promoción de la industria de los biosimilares, o ambas.
No en vano, en los Objetivos de Desarrollo del Milenio, ante las tasas diferenciales en materia de acceso a servicios de salud, que, por ejemplo, afecta significativamente a mujeres y a grupos étnicos, se estableció en el N.° 8 la meta 17 relacionada con la provisión de medicamentos esenciales a precios accesibles en los países en vías de desarrollo (45, 46).
En un modelo constitucional cuya legitimidad se basa en el respeto y la garantía de los derechos, y en la necesidad de remover los obstáculos que impidan su disfrute, seguramente no hace falta argumentación abundante sobre el fundamento ni sobre la obligación de garantizar la salud y las mejores condiciones de vida posibles, en particular cuando estas dependen del acceso a medicamentos. En este asunto, aunque no está exento de disputas sobre la medicalización y algunas de sus consecuencias, no parece ser el aspecto más problemático.
En cambio, sí presenta y expresa una relevante dificultad en materia de medicamentos y de las posibilidades de acceso a la información completa y comprensible de quienes los requieren. Por lo que, la protección de los consumidores de medicamentos, siguiendo la línea teórica revisada, se ha de ver desde la perspectiva del derecho al acceso a los medicamentos como derecho humano enmarcado en el derecho a la salud. Y el ejercicio de este último se efectiviza en la medida que este usuario, o el prestador de servicios, tenga los elementos necesarios para la toma responsable de decisiones relacionados con el consumo de estos, que debe apuntar a una garantía del CIM.
Conclusiones
Acorde con la información pública disponible del Invima, no todos los biosimilares autorizados por FDA y EMA se encuentran autorizados en Colombia, pero tampoco los biosimilares autorizados por el Invima lo están en estas dos agencias reguladoras. Adicionalmente, no existe información clara disponible en bases públicas que dé cuenta de una clasificación entre biológicas de referencia y biosimilares que se comercializan en el país.
En el caso de estos biosimilares registrados ante el Invima, sus titulares tienen autorización para importar y vender, de lo que se entiende que en el país no existe una industria biotecnológica que por el momento genere competitividad en términos de producción de estos medicamentos, en buena parte porque se encuentra en proceso la plena aplicación de la normativa relacionada con la producción de biológicos en el país, lo cual puede incidir en el incremento del costo del medicamento.
En relación con el derecho al acceso efectivo a medicamentos biológicos por vía judicial, se encontró que siete de estos biológicos han sido autorizados al menos una vez por esta vía a los usuarios del sistema y han logrado un pronunciamiento de la Corte Constitucional o de la Corte Suprema de Justicia colombianas. En términos generales, ambas corporaciones han señalado la necesaria conexión entre el acceso a medicamentos que son vitales para el paciente y la protección efectiva de los derechos a la integridad, la salud y la vida digna.
Esta judicialización de la salud, y, específicamente, las tutelas por medicamentos como una de las causas más frecuente de litigio en salud, ha contribuido a las medidas de creación de un del plan de beneficios, que ha permitido incluir los biológicos de referencia y sus biosimilares, para resolver los tradicionales problemas del sistema y las tutelas por “No-pos” en relación con los altos recobros, los sobrecostos innecesarios para el sistema de salud. A su vez, esto ha permitido que el Ministerio ejerza un control de precios, entre ellos, para algunos medicamentos biológicos, con lo que se busca superar algunas de estas inequidades del sistema.
Finalmente, se debe mencionar que si bien los decretos reglamentarios han sido de gran avance en relación con el acceso a biosimilares y, en consecuencia, a sus biológicos pioneros, el sistema aún tiene barreras jurídicas, de información, disponibilidad y acceso que contribuyan a garantizar la protección y efectividad del derecho a la salud de la población. En este sentido, es evidente que no se puede hablar de un CIM biológicos y biosimilares en el país, en la medida que ni siquiera el usuario del sistema, o el prestador, tienen claridad respecto de los medicamentos que efectivamente se encuentran autorizados y circulan en el país dentro de esta categoría.
Los sistemas de información son incipientes y el acceso a estos no resulta amigable ni completo para el consumidor, lo que afecta el derecho a un CIM. En consecuencia, además de los esfuerzos por su regulación, se debe trabajar en la generación de información suficiente y adecuada sobre ellos.
Artículo final del proyecto de investigación “Regulación de medicamentos de origen biológico en Colombia”, desarrollado y financiado por la Universidad del Rosario, adscrito a las líneas 1. Derechos Emergentes y enfoque diferencial del Grupo de Investigación en Derechos Humanos, 2. Geopolíticas del conocimiento del Grupo de Investigación en Derecho Público y 3. Propiedad Intelectual del Grupo de Derecho Privado. Facultad de Jurisprudencia, Universidad del Rosario.
Agradecimientos
Agradecemos de manera especial al Observatorio del Medicamento por la información suministrada en relación con los datos de los medicamentos analizados. Así mismo a los estudiantes Manuel Alejandro Rosero Castillo y Daniel Felipe Gaitán Peña, integrantes de la línea de investigación creada en el marco del proyecto “Regulación de Medicamentos de origen biológico en Colombia” y estudiantes del programa de Jurisprudencia de la Universidad del Rosario.
Referencias
1. Patel PK, King CR, Feldman SR. Biologics and biosimilars. J Dermatolog Treat. 2015;26(4):299-302. doi:10.3109/09546634.2015.1054782
2. Ahmed I, Kaspar B, Sharma U. Pharmacotherapy: Biosimilars: Impact of Biologic Product Life Cycle and European Experience on the Regulatory Trajectory in the United States. Clin Ther. 2012;34:400-19. doi: 10.1016/j.clinthera.2011.12.005
3. Gaviria A, González CPV, Muñoz CG, Morales ÁA. The debate on regulating biotechnology drugs: Colombia in the international context. Rev Panam Salud Publica; 2016 40(1):40-7.
4. Colombia, Ministerio de Salud y Protección Social. Decreto 1782 de 2014, por el cual se establecen los requisitos y el procedimiento para las Evaluaciones Farmacológica y Farmacéutica de los medicamentos biológicos en el trámite del registro sanitario (2014 sep 18). Disponible en: http://vlex.com/vid/evaluaciones-farmaca-utica-tra-mite-531084770
5. Díaz Vera LM, Herrera Sierra LF. Patentes de Medicamentos: Incentivos a la Innovación o Límites a la Salud Humana - Un análisis desde la propiedad intelectual y el derecho de la competencia. La propiedad inmaterial. 2013;(17):31-62.
6. Pinzón Camargo MA. Acceso a medicamentos y propiedad intelectual: un conflicto de derechos. Con-Texto Revista de Derecho y Economía. 2010;31:133-52.
7. Paul P, Mukhopadhyay K. Growth via intellectual property rights versus gendered inequity in emerging economies: An ethical dilemma for international business. J Bus Ethics. 2010;91(3):359-78. doi: 10.1007/s10551-009-0088-y
8. Núñez L, Rojas LE, Campo S, Castro JA. Retos y contradicciones de la industria biotecnológica actual. Biotecnol Apl.. 2007;24(3-4):7.
9. Rover MRM, Vargas-Pelaez CM, Rocha Farias M, Nair Leite S. Access to high-cost drugs in Brazil from the perspective of physicians, pharmacists and patients. Gac Sanit. 2015; 30(2):110-6. doi: 10.1016/j.gaceta.2015.12.005
10. Vélez-Arango AL, González-López JR. El acceso a medicamentos y su alcance por la vía judicial: análisis comparativo Colombia-España. Rev Fac Nac Salud Pública, 2015;33(1):121.
11. Dorado-Torres DF. La constitucionalización del acceso a los medicamentos y su relación con la propiedad intelectual: Reflexiones sobre el caso ecuatoriano. Rev Jurídicas. 2016; 13(2):85-99. doi: 10.17151/jurid.2016.13.2.7
12. Costa Chaves G, Fogaça Vieira M, Reis R. Acesso a medicamentos e propriedade intelectual no Brasil: reflexões e estratégias da sociedade civil. Sur Rev Int Direitos Humanos. 2008;5(8):170-98. doi: 10.1590/S1806-64452008000100009
13. Vaca C. La declaración de interés público de imatinib (Glivec®): entre lo dulce y lo amargo. Rev Univ Ind Santander Salud. 2016;(3):273 -4.
14. Velásquez Arango G. El acceso a medicamentos y la propiedad intelectual: contribución de la Organización Mundial de la Salud. Med Rev Medicina. 2014;36(1):56-78.
15. Uribe Arbeláez M . Impatentabilidad de medicamentos esenciales. La propiedad inmaterial, 2014;(18):55-84.
16. Biosimilars ECPG on MA and U of. A consensus information paper. What you need to know about biosimilar medicinal products. Brussels: European Commission [internet]. 2013 [2017 dic 17]. Disponible en: http://www.medicinesforeurope.com/wp-content/uploads/2016/03/biosimilars_report_en.pdf
17. Hardon A, Hodgkin C, Fresle D. Cómo investigar el uso de medicamentos por parte de los consumidores. Ginebra: Organización Mundial de la Salud [internet] 2004 [2015 may 04]. Disponible en: http://www.who.int/medicines/publications/WHOEDMPAR2004.2spa.pdf
18. Pombo ML, Di Fabio JL, Cortés MdlA. Review: Review of regulation of biological and biotechnological products in Latin American and Caribbean countries. Biologicals. 2009;37:271-6.
19. Paradise J. The Devil Is in the Details: Health-Care Reform, Biosimilars, and Implementation Challenges for the Food and Drug Administration. Jurimetrics. 2011;279-92.
20. Whitmore E. Development of FDA-regulated Medical Products: A Translational Approach. Milwaukee. Wisconsin: Quality Press; 2012.
21. Centers for Disease Control and Services, Department of Health and Human Prevention. Food and drug administration acts: federal food, drug and cosmetic act, as amended january 1980 public health service act, biological products radiation control for health and safety act fair packaging and labeling act. 1980 [internet]. [2017 feb 07] Disponible en: http://pesquisa.bvsalud.org/portal/resource/pt/mis-14207?lang=en
22. Ranjani N. Regulatory considerations for biosimilars. Perspect Clin Res. 2010;1(1):11-14.
23. Europa. Parlamento Europeo y del Consejo. Directiva 2001/83/CE, por la que se establece un código comunitario sobre medicamentos para uso humano (2011 nov 6). Diario Oficial 2001;l(311):0067-128.
24. European Medicines Agency. Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical issues. London: EMA [internet]. 2006 [2017 feb 08]. Disponible en: http://www.EMA.europa.eu/docs/en_GB/document_library/Scientific_guideline/2015/01/WC500180219.pdf
25. US Food and Drug Administration. Purple Book: lists of licensed biological products with reference product exclusivity and biosimilarity or interchangeability evaluations. Silver Spring: FDA [internet] 2016 [2017 dic 13]. Disponible en: https://www.FDA.gov/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/TherapeuticBiologicApplications/Biosimilars/ucm411418.htm
26. Agency EM. Biosimilar medicines. En: Medicines Rfb, ed. Excel. London: EMA; 2016.
27. Rengifo García E. Propiedad intelectual el moderno derecho de autor. 2a ed. Bogotá: Universidad Externado de Colombia; 1999.
28. Ascarelli T. Teoría de la concurrencia y de los bienes inmateriales (traducción de E. Verdera y L. Suárez-Llanos),(1º edic.). Bolonia: Publicaciones del Real Colegio de España; 1970.
29. Comunidad Andina de Naciones. Régimen Común sobre Propiedad Industrial, Stat. Decisión 486 de 14 de septiembre de 2000 (2000 sept 19) [internet]. [2016 oct 10] Disponible en: https://app-vlex-com.ez.urosario.edu.co/#WW/search/*/decision+486/WW/vid/336233089
30. Heinemann A. Propiedad intelectual. En: Propiedad intelectual Reflexiones [internet]. 2012 [citado 2013 mar 05]. Disponible en: http://vlex.com/vid/propiedad-intelectual-426372722
31. Metke Méndez R. Lecciones de propiedad industrial. Bogotá: Baker & MacKenzie; 2001.
32. Fernández de Córdova M. Las licencias obligatorias de patentes en el Ecuador. Una breve referencia al caso de los medicamentos y al decreto presidencial 118. Iuris Dictio. 2013;15:205-20.
33. Botana-Agra JM. Invención y Patente. En: Fernández-Novoa C, Otero Lastres JM, Botana-Agra M, eds. Manual de la Propiedad Industrial. Madrid: Marcial Pons; 2017. pp. 99-112.
34. Super Intendencia de Industria y Comercio. Banco de Patentes Bogotá [internet]. 2011 [citado 2016 sep 08]. Disponible en: http://www.sic.gov.co/banco-de-patentes
35. Reyes Villamizar JL. Las reivindicaciones en las patentes de invención: su concepto, importancia y características principales. En: Metke Méndez R, León Robayo EI, Varela Pezzano E, eds. Propiedad Intelectual: Reflexiones. Bogotá: Editorial Universidad del Rosario; 2012. pp. 27-52.
36. Botana-Agra JM. Los certificados complementarios de protección de medicamentos y de productos fitosanitarios. En: Fernández-Nóvoa C, Otero Lastres JM, Botana-Agra JM, eds. Manual de la propiedad industrial. Madrid: Marcial Pons; 2017. pp. 307-318.
37. Pons del Valle Alomar M. Los límites de las patentes farmacéuticas. Anuario Facultad de Derecho de la Universidad de Alcalá. 2008;1:273-88.
38. Colombia, Ministerio de Salud. Decreto 677 de 1995, Por el cual se reglamenta parcialmente el Régimen de Registros y Licencias, el Control de Calidad, así como el Régimen de Vigilancia Sanitaria de Medicamentos, Cosméticos, Preparaciones Farmacéuticas a base de Recursos Naturales, Productos de Aseo, Higiene y Limpieza y otros productos de uso doméstico y se dictan otras disposiciones sobre la materia. (1995 abr 06). Disponible en: https://www.invima.gov.co/images/pdf/medicamentos/decretos/decreto_677_1995.pdf
39. Instituto Nacional de Vigilancia de Medicamentos y Alimentos. Trámite registros sanitario de medicamentos de síntesis quimia y biológicos 2010 [citado 2016 sept 08]. Disponible en: https://www.invima.gov.co/index.php/tramites-y--servicios/tr%C3%A1mites.html#informaci%C3%B3n-asociada
40. Villamañán E, González D, Armada E, Ruano M, Álvarez-Sala R, Herrero A. Juego de patentes. Sobre medicamentos genéricos y biosimilares. Rev Calid Asist. 2016;31(2):99-105.
41. Consejo Nacional de Política Económica y Social, Departamento Nacional de Planeación. Documento Conpes Social 155, Política Farmacéutica Nacional. Bogotá, [internet] 2012 [citado 2017 feb 09]. Disponible en: https://www.minsalud.gov.co/Documentos%20y%20Publicaciones/Politica%20Farmac%C3%A9utica%20Nacional.pdf
42. Rathore AS. Biosimilars in India. J Proteomics. 2015;127, Part A:71-2. https://doi.org/10.1016/j.jprot.2015.08.005
43. A. VVSS. Guía farmacológica. Madrid: Vidal Vademecum Spain S.A; 2016.
44. Fernández-Novoa C, Otero Lastres JM, Botana Agra JM. Hacia un nuevo sistema de patentes. Madrid: Ed. Montecorvo; 1982.
45. Ferreira da Silva MJ. Objetivos de Desarrollo del Milenio (ODM): el hambre, la salud de las mujeres y de los niños en Latinoamérica. Rev Cienc Salud. 2008;6(2):5-8.
46. Robles Llamazares M. Objetivos de Desarrollo del Milenio. Humanismo y Trabajo Social. 2012;(5):93-101.
Notas
4
“Decreto 1782 de 2014. Art. 6. Requisitos de Información común a las tres rutas. Independientemente de la ruta, el solicitante deberá presentar la siguiente información imprescindible, referida al medicamento objeto de la solicitud:
6.1. Descripción detallada del proceso y lugar de producción
6.2. Sistema de expresión
6.3. Pruebas de identidad biológica
6.4. Evaluación de la potencia
6.5. Propiedades fisicoquímicas
6.6. Evaluación de la actividad biológica
6.7. Evaluación de la pureza
6.8. Pruebas de inmunogenicidad
6.9. Plan de gestión de riesgo” (4).
5
Datos tomados de la List of Licensed Biological Products with (1) Reference Product Exclusivity and (2) Biosimilarity or Interchangeability Evaluations to Date actualizada el 13 de diciembre de 2017 en el Purple Book que se refiere a la lista de medicamentos biológicos, incluyendo los biosimilares, aprobados por el Center for Drug Evaluation and Research (CDER).
6
Datos tomados del European Public Assessment Reports publicado por la EMA y actualizado a enero 20 de 2018.
7
Según el artículo 9.º de la Decisión 486 de 2000, una reivindicación de prioridad consiste en “la primera solicitud de patente de invención o de modelo de utilidad, o de registro de diseño industrial o de marca, válidamente presentada en otro País Miembro o ante una autoridad nacional, regional o internacional con la cual el País Miembro estuviese vinculado por algún tratado que establezca un derecho de prioridad análogo al que establece la presente Decisión, conferirá al solicitante o a su causahabiente un derecho de prioridad para solicitar en el País Miembro una patente o un registro respecto de la misma materia” (29).
8
Esta expresión no encuentra una definición única en los sistemas legales del mundo, razón por la cual es entendida como una solución nueva a un problema técnico. Así lo explica Metke, quien al analizar la Decisión 486 de la can no encuentra una definición de este término, lo cual justifica para evitar el riesgo de que se limite el concepto, “que continuamente está en evolución por los avances de la técnica, lo que a su vez limitaría la facultad de apreciación de los examinadores al emitir los juicios sobre la patentabilidad de la invención” (31).
9
En Colombia es conocido como Centro de Información Tecnológica y Apoyo a la Gestión de la Propiedad Industrial de la Superintendencia de Industria y Comercio. Esta dependencia permite conocer la información sobre los documentos de patentes y modelos de utilidad publicados a escala nacional o internacional (33).
10
Cuando no está comprendida en el estado de la técnica, es decir, todo lo que haya sido accesible al público por una descripción escrita u oral, utilización, comercialización o cualquier otro medio antes de la fecha de presentación de la solicitud de patente o, en su caso, de la prioridad reconocida (29).
11
Si su objeto no es obvio para una persona normalmente versada en el arte respectivo o se desprende necesariamente del arte anterior (29).
12
Cuando su objeto puede ser desarrollado industrialmente en cualquier sector productivo, incluidos los servicios (29).
13
Es pertinente señalar que se presentó un derecho de consulta ante el Invima para acceder al listado oficial de medicamentos biológicos, biotecnológicos y biosimilares actualmente disponibles en el país. Esta solicitud se hizo por medio de la web y el correo electrónico de consultas del Invima en agosto de 2016, a la que se obtuvo como respuesta que para acceder a esta información debía hacerse un pago, a lo cual los investigadores dimos como respuesta que se indicara el trámite para el respectivo pago, sin que a la fecha se hubiese obtenido nueva comunicación por parte de la agencia reguladora.
14
Vigente a partir del 01 de marzo de 2018
15
Este medicamento ha sido polémico en el país desde su autorización por el Invima. En primer lugar, por las reclamaciones que en su momento presentó la Asociación Colombiana de Reumatología y Laboratorios Wyeth, y, en segundo lugar, por los derechos de petición y las tutelas presentadas por pacientes solicitando que se administrase en el ENBREL en lugar del ETANAR.
16
Este medicamento en la FDA se considera biológico original y no biosimilar.
17
No se referencia como un producto biosimilar, pero la compañía Winthrop Pharmaceuticals se define a sí misma como la línea de genéricos de Sanofi.
18
Laboratorio de la India.
19
Pertenece al grupo Intas Pharmaceutical Ltd. de la India, con dedicación especial a la fabricación de medicamentos genéticos y ahora biosimilares.
20
Tiene actualmente trámite ante la FDA para su aprobación.
21
Líder en genéricos y perteneciente al grupo Sandoz.
22
Se comercializa en Chile y Brasil.
23
Es una compañía de la Argentina de biosimilares.
24
Corresponde al mismo biosimilar leucostim, fabricado por la compañía koreana DONG-A ST CO., LTDA. Laboratorios Chalver tiene el rol de acondicionador e importador para el medicamento Leucosos.
25
Tiene derechos exclusivos de comercialización de los productos desarrollados por el Centro de Ingeniería Genética y Biotecnología de Cuba.
26
Es un laboratorio Uruguayo, y en Colombia comercializado a través de Scandinavia Pharma.
27
Laboratorio de la India. Comercializado en Colombia a través del Laboratorio Franco Colombiano Lafrancol S.A.S.
28
Laboratorio de la India. Comercializado en Colombia a través SICMAFARMA S.A.S. (importador) y Biothermics de Colombia S.A.
29
Laboratorio de la india. La titularidad del registro en Colombia es de BR PHARMA INTERNATIONAL S.A.S.
30
Mylan GmbH, presentó ante la EMA solicitud de autorización de comercialización del Ogivri, sin embargo, en agosto de 2017 notificó oficialmente al Comité de Medicamentos de Uso Humano (CHMP) de esta Agencia que deseaba retirar su solicitud de autorización de comercialización de Ogivri.
31
Este biosimilar, recibió aprobación de la FDA en diciembre de 2017. Sin embargo, aún no se encuentra incluido en la lista de biosimilares del Purple Book.
32
Se considera biológico original.
33
Farmacéutica de la India. En el año 2009, Sanofi envió un comunicado al Invima, señalando que este medicamento no cuenta con estudios de eficacia y seguridad. Ver: https://www.invima.gov.co/images/pdf/salas-especializadas/Sala_Especializada_de_Medicamentos/acta2009/acta332009_medicamentos.pdf
34
Farmaceútica de la China, importado por el Laboratorio Franco Colombiano.
35
Registro sanitario en trámite de renovación. Fabricado por Blau Farmaceutica S.A. de Brasil e importado por Blau Farmacéutica Colombia S.A.
36
El registro sanitario de este biosimilar es de titularidad de este laboratorio colombiano en la modalidad de fabricar y vender.
37
Laboratorio de la India. En Colombia comercializado por Willow Pharma S.A.S.
38
Fabricado por Amgen y financiado por J&J (Janssen - Cila). Actualmente se trata de un producto descontinuado, en principio por los efectos secundarios del medicamento. Sin embargo, existen en el mercado otros biológicos y/o biosimilares cuyo P.A. es epoetin alfa.
39
Los biosimilares Retacrit de Hospira UK Limited y Silapo de Stada Arzneimittel, autorizados por la EMA, se considera biosimilares de eprex/erypro, pero su principio activo es epoetin zeta.
40
De Brasil.
41
Pertenece al grupo empresarial cubano Biocubafarma.
42
De una farmacéutica Argentina, que lo considera un biosimilar.
43
Cubano, considerado un biogenérico (ver: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1164417/)
44
Retirado por su casa farmacéutica.
45
Biológico original. No es considerado producto de referencia.
46
Biológico original. No es considerado producto de referencia.
47
Biológico original. No es considerado producto de referencia.
48
Biológico original.
49
Precio Promedio de la Presentación en Sismed-PPPS.
50
Esta variación (%v) es entre el PPPS del biológico de referencia (P1) y PPSS del biosimilar (P2), para lo cual se aplicó la formula %V=(P1-P2)/P1.
51
La doctrina plantea además otras situaciones como: (i) destinación exagerada de recursos personales y económicos para la organización, gestión y mantenimiento del sistema por parte del Gobierno; (ii) paralización en el proceso de búsqueda y generación de ulteriores invenciones, lo que conlleva a que el titular de la patente no busque obtener nuevas creaciones, y (iii) perpetuación de la exclusividad mediante la agregación de ulteriores patentes obtenidas en la propia empresa o a través de la adquisición de patentes a terceros (33).
52
Regulada en los artículos 61 y siguientes de la Decisión 486, conforme a los cuales: “Vencido el plazo de tres años contados a partir de la concesión de la patente o de cuatro años contados a partir de la solicitud de la misma, el que resulte mayor, la oficina nacional competente, a solicitud de cualquier interesado, otorgará una licencia obligatoria principalmente para la producción industrial del producto objeto de la patente o el uso integral del procedimiento patentado, sólo si en el momento de su petición la patente no se hubiere explotado en los términos que establecen los artículos 59 y 60, en el País Miembro donde se solicite la licencia, o si la explotación de la invención hubiere estado suspendida por más de un año” (29).
53
Conocida en Estados Unidos como la excepción Hatch Waxman, exonera de la violación de derechos derivados de las patentes todos los usos de los compuestos relacionados con la provisión de información al gobierno bajo cualquier ley que regula la fabricación, el uso o la distribución de drogas.
Notas de autor
* Diana
Rocío Bernal-Camargo, PhD
Autora
de correspondencia: diana.bernalc@urosario.edu.co
** Julio
César Gaitán-Bohórquez, PhD
*** Édgar
Iván León-Robayo, PhD