
10.12804/revistas.urosario.edu.co/revsalud/a.13637
ARTÍCULOS DE INVESTIGACIÓN CLÍNICA O EXPERIMENTAL
Manuel Huertas-Quiñones 1
Fernando Suárez-Obando 2
Norma Carolina Barajas-Viracachá 3
Paulo César Becerra-Ortiz 4
Edna Julieth Bobadilla-Quesada 5
Carlos Ernesto Bolaños-Almeida 6
José Manuel Cañón-Zambrano 7
Sandra Milena Castellar-Leones 8
Jenny Libeth Jurado-Hernández 9
Juan David Lasprilla-Tovar 10
Nicolás J. Laza-Gutiérrez 11
Isabel C. Londoño Ossa 12
Blair Ortiz-Giraldo 13
Fernando Ortiz-Corredor 14
Sandra Janeth Ospina-Lagos 15
Juan Carlos Prieto 16
Carolina Rivera-Nieto 17
Edicson Ruiz-Ospina 18
Felipe Ruiz-Botero 19
María Claudia Salcedo-Maldonado 20
Diana Pilar Soto-Peña 21
Lina Marcela Tavera-Saldaña 22
María Julia Torres-Nieto 23
Diana Carolina Sánchez-Peñarete 24
1 Departamento de Cardiología Pediátrica. Coordinador de la Clínica de Falla Cardíaca y
Trasplante Cardíaco Pediátrico, Instituto de Cardiopatías Congénitas, Fundación CardioInfantil-Instituto de Cardiología (Bogotá, Colombia).
Profesor titular de la Universidad Nacional de Colombia (Bogotá, Colombia).
 0000-0002-2552-9870
0000-0002-2552-9870
 manuelhuertasmd@gmail.com
manuelhuertasmd@gmail.com
2 El Dr. Suárez falleció posteriormente a su participación como autor de este artículo. En ese momento, era el director del Instituto de Genética Humana, Pontificia Universidad
Javeriana (Bogotá, Colombia). Perteneció al Departamento de Genética, Hospital Universitario San Ignacio (Bogotá, Colombia).
0000-0001-6336-5347
3 Neuróloga infantil, Fundación Cardiovascular de Colombia-Hospital Internacional de Colombia (Piedecuesta, Santander, Colombia).
0000-0002-9494-2618
4 Departamento de Medicina Física y Rehabilitación, Somefyr S. A. S. (Cúcuta, Colombia).
0009-0003-0599-5633
5 Departamento de Neurología Infantil, Hospital Universitario San Ignacio (Bogotá, Colombia).
Unidad de Neuropediatría, Junta de Enfermedades Neuromusculares, Fundación HOMI (Hospital Pediátrico
La Misericordia) (Bogotá, Colombia).
0000-0002-5023-4644
6 Coordinador del Laboratorio de Sueño, Unidad de Neuropediatría, Fundación HOMI (Hospital Pediátrico La Misericordia) (Bogotá, Colombia).
0000-0002-0064-723X
7 Neurólogo pediatra del Instituto Neurológico de Colombia (Medellín, Colombia).
0000-0002-3709-0500
8 Miembro de la Junta de Enfermedades Neuromusculares de Biotecgen.
Profesor del Departamento de Medicina Física y Rehabilitación, Facultad de Medicina, Universidad Nacional de Colombia.
Proceso de Rehabilitación del Hospital Universitario Nacional de Colombia (Bogotá, Colombia).
0000-0002-4559-2965
9 Subdirectora de Docencia e Investigación Científica, Hospital Militar Central (Bogotá,
Colombia). Neumóloga pediatra y especialista en Docencia Universitaria.
0000-0002-6610-9207
10 Endocrinólogo pediatra de la Fundación HOMI (Hospital Pediátrico La Misericordia) (Bogotá, Colombia).
Endocrinólogo pediatra, Clínica Marly Jorge Cavelier Gaviria (Bogotá, Colombia).
0000-0002-7165-0057
11 Departamento de Neurología Infantil, NeuroXtimular S. A. S. IPS (Barranquilla, Colombia).
0009-0007-1255-8553
12 Especialista en Medicina Física y Rehabilitación. Docente de Rehabilitación Pediátrica, Universidad del Valle (Cali, Colombia).
Miembro de la Junta de Enfermedades Neuromusculares de la Fundación Clínica Infantil Club Noel (Cali, Colombia).
Miembro de la Junta de Enfermedades Neuromusculares del Hospital Universitario del Valle (Cali, Colombia).
0000-0002-9902-3514
13 Departamento de Neurología Infantil, Hospital San Vicente Fundación (Medellín, Colombia).
0000-0002-9165-4004
14 Profesor de la Universidad Nacional de Colombia (Bogotá, Colombia).
Jefe del Departamento de Medicina Física y Rehabilitación.
0000-0002-7427-3576
15 Genetista. Profesora asistente de la Universidad Nacional de Colombia (Bogotá, Colombia).
0000-0003-0397-0910
16 Profesor asistente del Instituto de Genética Humana, Pontificia Universidad Javeriana (Bogotá, Colombia).
0000-0001-8706-0775
17 Jefe del Servicio de Genética Médica, Hospital Pediátrico-Fundación CardioInfantil (Bogotá, Colombia).
0000-0002-8853-6509
18 Profesor de Medicina Física y Rehabilitación de la Universidad Nacional de Colombia (Bogotá, Colombia).
Medico fisiatra de la Fundación HOMI (Hospital Pediátrico de La Misericordia) (Bogotá, Colombia).
0000-0002-3664-4903
19 Profesor de la Facultad de Ciencias de la Salud e investigador del Centro de Investigaciones en Anomalías Congénitas y Enfermedades Raras, Universidad Icesi (Cali,
Colombia).
0000-0001-9536-7080
20 Fisiatra. Miembro de la Junta de Enfermedades Neuromusculares del Instituto Roosevelt (Bogotá, Colombia).
0000-0003-3207-4701
21 Coordinadora del Departamento de Fisioterapia, Instituto Roosevelt (Bogotá, Colombia).
0000-0003-0401-1404
22 Neuróloga pediatra. Directora científica de Neuroconexión IPS (Armenia, Colombia).
Profesora titular de la Universidad del Quindío (Armenia, Colombia).
0000-0002-6589-0249
23 Neurología infantil. Consultorio particular (Valledupar, Colombia).
0009-0003-1567-3451
24 Junta de Enfermedades Neuromusculares del Instituto Roosevelt (Bogotá, Colombia).
0000-0003-4612-3324
Recibido: 1 de febrero de 2024
Aprobado: 16 de enero de 2025
Para citar este artículo: Huertas-Quiñones M, Suárez-Obando F, Barajas-Viracachá NC, Becerra-Ortiz PC, Bobadilla-Quesada EJ, Bolaños-Almeida CE, Cañón-Zambrano JM, Castellar-Leones SM, Jurado-Hernández JL, Lasprilla-Tovar JD, Laza-Gutiérrez NJ, Londoño Ossa IC, Ortiz-Giraldo B, Ortiz-Corredor F, Ospina-Lagos SJ, Prieto JC, Rivera-Nieto C, Ruiz-Ospina E, Ruiz-Botero F, Salcedo Maldonado MC, Soto-Peña DP, Tavera-Saldaña LM, Torres-Nieto MJ, Sánchez-Peñarete DC. Diagnóstico y manejo de las complicaciones cardíacas en el paciente con distrofia muscular de Duchenne. Rev Cienc Salud. 2025;23(esp.):1-15. https://doi.org/10.12804/revistas.urosario.edu.co/revsalud/a.13637
Resumen
La distrofia muscular de Duchenne es la distrofia muscular más frecuente y severa en la infancia. A medida que progresa la enfermedad, se evidencia afectación del músculo cardíaco, con la presencia de miocardiopatía dilatada y miocardiopatía hipertrófica o restrictiva que llevan a falla cardíaca. Actualmente, no se cuenta con un tratamiento curativo para la distrofia muscular de Duchenne o sus complicaciones cardiovasculares. Sin embargo, el uso de glucocorticoides, la rehabilitación física, la ventilación mecánica no invasiva y el manejo multidisciplinario con un enfoque cardiorrespiratorio y ortopédico han mostrado modificar la historia natural de la enfermedad. El presente artículo proporciona una síntesis sobre el diagnóstico y tratamiento de los pacientes con diagnóstico de distrofia muscular de Duchenne que presentan complicaciones cardiovasculares.
Palabras clave: Duchenne; cardíaco; cardiomiopatía hipertrófica; cardiomiopatía dilatada.
Abstract
Duchenne muscular dystrophy is the most common and severe muscular dystrophy in childhood. As the disease progresses, there is evidence of cardiac muscle involvement with the presence of dilated cardiomyopathy and hypertrophic or restrictive cardiomyopathy, leading to heart failure. Currently, there is no curative treatment for Duchenne muscular dystrophy or its cardiovascular complications. However, the use of glucocorticoids, physical rehabilitation, non-invasive mechanical ventilation, and multidisciplinary management with a cardiorespiratory and orthopedic focus have been shown to modify the natural history of the disease. This article provides a synthesis of the diagnosis and treatment of patients diagnosed with Duchenne muscular dystrophy who present cardiovascular complications.
Keywords: Duchenne; cardiac; hypertrophic cardiomyopathy; dilated cardiomyopathy.
Resumo
A distrofia muscular de Duchenne é a distrofia muscular mais comum e grave na infância. À medida que a doença progride, há evidências de envolvimento do músculo cardíaco com a presença de cardiomiopatia dilatada e cardiomiopatia hipertrófica ou restritiva levando à insuficiência cardíaca. Atualmente, não há tratamento curativo para a distrofia muscular de Duchenne ou suas complicações cardiovasculares. No entanto, o uso de glicocorticoides, a reabilitação física, a ventilação mecânica não invasiva e o manejo multidisciplinar com abordagem cardiorrespiratória e ortopédica demonstraram modificar a história natural da doença. Este artigo fornece uma síntese do diagnóstico e tratamento de pacientes com diagnóstico de distrofia muscular de Duchenne que apresentam complicações cardiovasculares.
Palavras-chave: Duchenne; cardíaco; cardiomiopatia hipertrófica; cardiomiopatia dilatada.
Introducción
En los pacientes con distrofia muscular de Duchenne (DMD) hay manifestaciones cardiovasculares derivadas de la afectación muscular que produce el avance de la enfermedad (1). Aproximadamente un 90 % de los pacientes con DMD presentan miocardiopatía dilatada por alteraciones de la distrofina cardíaca y, en menor frecuencia, miocardiopatía hipertrófica o miocardiopatía restrictiva (1-3). Uno de los principales síntomas cardíacos en los pacientes con DMD es la falla cardíaca. Se trata de un síndrome fisiopatológico y clínico progresivo en el que el paciente presenta signos característicos asociados con trastornos circulatorios, neurohormonales y moleculares (1-3). Este síndrome se relaciona también con alteraciones cardiovasculares y extracardíacas, como edema, dificultad respiratoria, retraso ponderal e intolerancia al ejercicio (4,5).
A la fecha no existe un tratamiento curativo para la DMD ni para sus complicaciones cardiovasculares. Sin embargo, como resultado de la disponibilidad de la terapia con glucocorticoides, de la rehabilitación física, de la ventilación mecánica no invasiva y del manejo multidisciplinario con un enfoque cardiorrespiratorio y ortopédico, se ha logrado modificar la historia natural de la enfermedad (6-8) y mejorar la sobrevida de estos pacientes.
Fisiopatología de las manifestaciones cardíacas de la distrofia muscular de Duchenne
La función de la distrofina es ligar el citoesqueleto interno a la matriz extracelular; de ahí que desempeñe un importante papel en la estabilización celular (9). Al ligar el componente intracelular (actina) con el complejo glicoproteico de la membrana celular, proporciona soporte mecánico durante la contracción muscular (1,10).
La alteración en la estructura de la distrofina, secundaria a las mutaciones asociadas con la DMD, afecta la unión entre el citoesqueleto y la matriz extracelular, con posterior debilitamiento del sarcolema y la degeneración celular progresiva (1). En el corazón, la degeneración de los cardiomiocitos lleva a fibrosis (11). Inicialmente, este fenómeno aparece en la pared inferolateral y perjudica progresivamente el ventrículo izquierdo, hacia el final de la segunda década de la vida (3). Conforme aumenta la fibrosis miocárdica, hay una dilatación progresiva del ventrículo izquierdo con el respectivo incremento del trabajo cardíaco, activación del sistema renina-angiotensina y del sistema nervioso simpático (3).
Ante la alteración en la arquitectura del ventrículo izquierdo o del ventrículo derecho, cambia la remodelación ventricular (los cardiomiocitos son remplazados por fibroblastos), disminuye la función cardíaca y aparecen focos arritmogénicos (6). Además, las arritmias deterioran aún más el cuadro de falla cardíaca y se crea un círculo vicioso entre estas dos patologías. Adicionalmente, estos pacientes presentan taquicardia asociada con la disfunción del sistema nervioso autónomo. Esta falta de sincronía del ventrículo izquierdo, vinculada con una alta frecuencia cardíaca secundaria a la disfunción del sistema nervioso autónomo, contribuye a empeorar progresivamente la disfunción del ventrículo izquierdo (3).
Manifestaciones cardíacas en la distrofia muscular de Duchenne
En estadios iniciales, el compromiso cardiovascular en pacientes con DMD puede pasar desapercibido, a pesar de la disfunción cardíaca. Es más, hay un alto porcentaje de pacientes asintomáticos debido a la ausencia de actividad física por la pérdida de la deambulación (4,7,8,12). Las manifestaciones son variadas y progresivas, y se hacen notorias a los 10 años, entre las cuales la alteración de la función diastólica es el síntomas más frecuente (3,13).
Como consecuencia de los cambios en la arquitectura del corazón, el ventrículo izquierdo pierde el movimiento de torsión. La fracción de eyección se reduce en aproximadamente el 50 % que provoca una disfunción sistólica importante con hipertensión pulmonar secundaria (5,13,14). La miocardiopatía no compactada se reporta en el 28 % de los pacientes con DMD y se asocia con disminución en la función sistólica del ventrículo izquierdo, trastornos del ritmo cardíaco, falla cardíaca y eventos cerebrovasculares (13). En un menor porcentaje de pacientes, se evidencia miocardiopatía hipertrófica con disfunción diastólica temprana (15,16). A medida que avanza la cardiomiopatía, en los pacientes se producen un síndrome de bajo gasto cardíaco (signos de hipoperfusión sistémica, acidosis metabólica o mala perfusión) y un síndrome de congestión venosa sistémica o congestión venosa pulmonar, que rápidamente se convierten en una falla cardíaca global (12).
Clase funcional en falla cardíaca
La valoración de la clase funcional es compleja en pacientes con DMD, debido a que los factores evaluados no se encuentran validados para estos pacientes (17). Esto hace que las clasificaciones disponibles no sean las más adecuadas para hacer el seguimiento clínico de la clase funcional en los pacientes con DMD. En adolescentes y adultos es posible usar la clasificación de la New York Heart Association (NYHA) o la escala de la American College of Cardiology/American Heart Association Task Force (ACC/AHA) (18-20), sin que sean específicas para pacientes con DMD. En niños también es posible el uso de la escala de clasificación de Ross, que determina una valoración global de la severidad de la falla cardíaca, aunque esta escala tampoco es específica para pacientes con DMD (21,22) (tabla 1). En general, se puede utilizar la clasificación por estadios ACC-AHA en estos pacientes (tabla 2) (23).
Tabla 1. Clasificación de NYHA y clasificación de Ross
Clase |
New York |
|
I |
• No limitaciones en la actividad física |
• Asintomático |
II |
• Ligera limitación de la actividad física |
• Taquipnea leve o sudoración con la alimentación en lactantes |
III |
• Limitación marcada de la actividad física |
• Marcada taquipnea o sudoración con la alimentación en lactantes |
IV |
• Incapacidad de cualquier actividad física sin síntomas |
• Síntomas en reposo: taquipnea, sudoración, retracciones |
Tabla 2. Clasificación por estadios según el American College of Cardiology y la American Heart Association (20)
Estadio A |
Estadio B |
Estadio C |
Estadio D |
Fuente: tomado y adaptado de Spurney CF. Cardiomyopathy of Duchenne muscular dystrophy: current understanding and future directions. Muscle Nerve. 2011;44(1):8-19.
Evaluación de las manifestaciones cardíacas y seguimiento en la distrofia muscular de Duchenne
Dada la variabilidad del espectro clínico y las difusas manifestaciones en pacientes con DMD, es necesario un diagnóstico temprano que establezca una terapia precoz y adecuada, así como intervenciones antes de que aparezca la insuficiencia cardíaca (4,7,8,12,25). Esto es fundamental para mejorar la calidad de vida y maximizar la supervivencia de los afectados. El objetivo es entonces abordar tempranamente a los pacientes con DMD, en el estadio preclínico, para poder determinar por ecocardiografía, Strain, resonancia, electrocardiograma y biomarcadores, la presencia de disfunción diastólica (12,24-27).
En principio, se recomienda una evaluación cardiovascular integral, en el momento del diagnóstico de DMD, que incluya valoración clínica por cardiología pediátrica, electrocardiograma basal, imágenes no invasivas como ecocardiografía (examen de elección en estos pacientes) y resonancia magnética nuclear cardíaca, que permiten evaluar la viabilidad miocárdica, determinar zonas de fibrosis y evaluar puntajes de riesgo (12,24,25,27). Sin embargo, pueden presentarse limitaciones para realizar estos exámenes relacionadas con su costo y disponibilidad (20,28). Nuevas técnicas ecocardiográficas como el Strain permiten detectar tempranamente cambios mínimos en la función sistólica y diastólica, incluso por debajo de las edades establecidas (29,30).
El seguimiento del paciente con DMD debe ser integral, óptimo y multidisciplinario, con recomendaciones que varían de acuerdo con la fase en la que se encuentra (tabla 3). Se requiere la toma de exámenes paraclínicos y estudios imagenológicos, entre los cuales se encuentran (31-34):
1. Laboratorios: hemograma, proteína C reactiva, enzimas cardíacas (troponina), electrolitos, perfil de infección, cistatina C (un marcador de función renal no influenciado por la degradación del músculo esquelético) y pruebas de función hepática (que pueden estar alteradas por congestión venosa sistémica).
2. Biomarcadores: para el seguimiento de los pacientes, se recomienda la toma de péptido natriurético tipo B (su uso debe ser racional e individualizado). Valores >300 pg/ml son un predictor de hospitalización por insuficiencia cardíaca o mal pronóstico (20).
3. Radiografía de tórax: para definir alteraciones propias de la enfermedad de base o comorbilidades.
4. Electrocardiograma: puede evidenciar un ritmo sinusal con algunos trastornos en la repolarización o aumento en la amplitud de la onda R, extrasístoles ventriculares o supraventriculares y salvas de taquicardia ventricular (35).
5. Ecocardiografía: debe incluir fracción de eyección, fracción de acortamiento, desplazamiento sistólico del plano del anillo tricúspideo, examen Doppler pulsátil, continuo y tisular, y el uso de Strain, para medir la deformación en los distintos segmentos cardíacos y evaluar mejor la función cardíaca.
Tabla 3. Seguimiento y diagnóstico de pacientes con distrofia muscular de Duchenne (34)
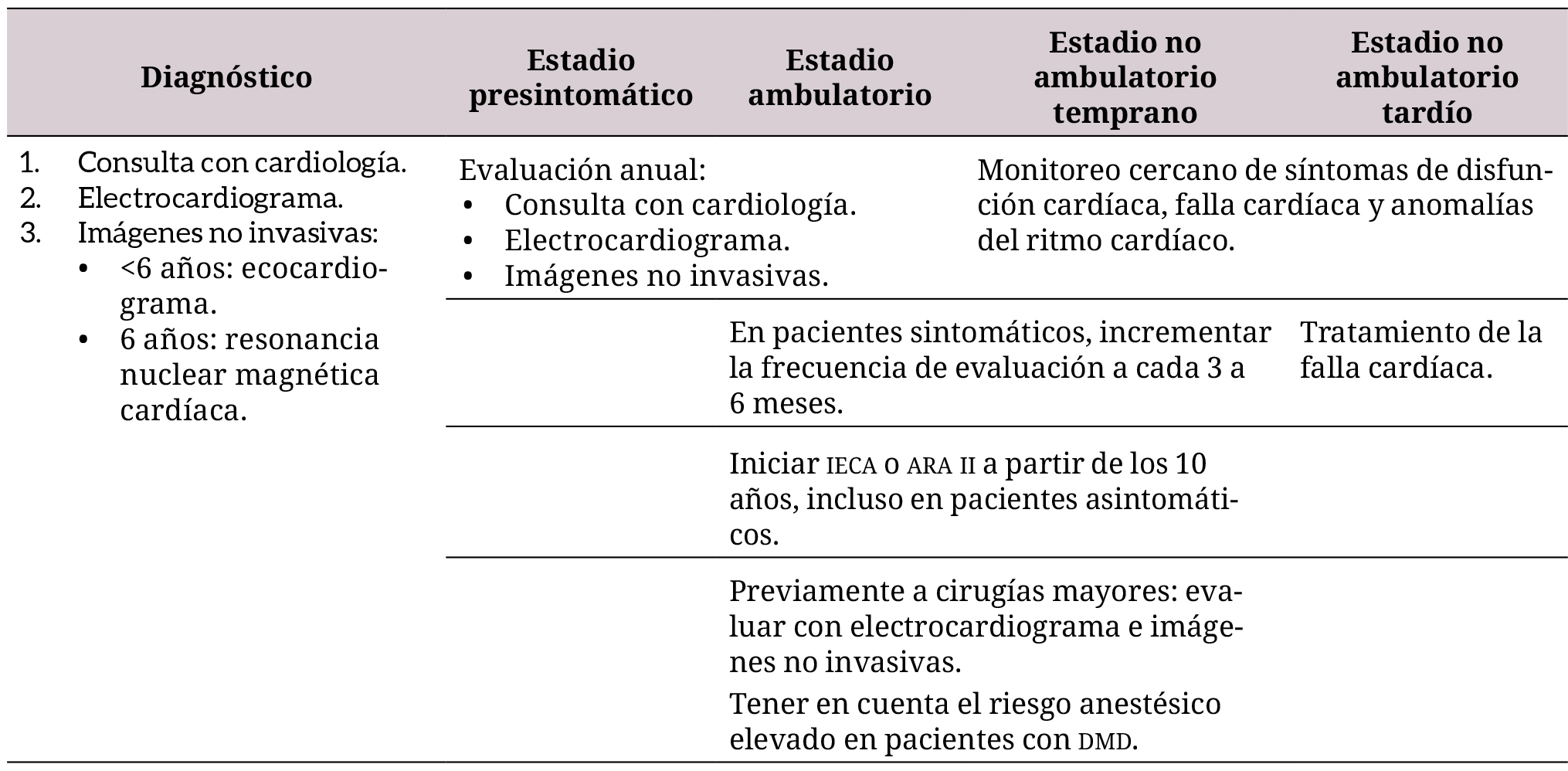
IECA: enzima convertidora de la angiotensina;
ARA II: antagonista de los receptores de la angiotensina n.
En pacientes con DMD asintomáticos desde el punto de vista cardiovascular, se recomienda la toma de un ecocardiograma al diagnóstico y hacia los 6 años de vida, con un seguimiento anual hasta los 10 años. Luego, según la afectación cardiovascular, los controles deben ser más frecuentes (cada 3 a 6 meses), con evaluaciones clínicas y paraclínicas (incluyendo electrocardiograma, ecocardiograma, Holter y resonancia magnética nuclear cardíaca) que permitan identificar el momento ideal para la intervención y el establecimiento del tratamiento (25,28,34,36,37).
Tratamiento de las manifestaciones cardíacas en la distrofia muscular de Duchenne (23,27,31)
Las terapias establecidas para tratar los efectos musculares de la enfermedad en pacientes con DMD son inefectivas para tratar el compromiso cardiovascular (27,28,34). El manejo farmacológico de la insuficiencia cardíaca se basa en diuréticos, inhibidores de la enzima convertidora de la angiotensina y betabloqueadores, de acuerdo con el estadio del paciente (tabla 4) (5,28,38).
Tabla 4. Tratamiento de la insuficiencia cardíaca en pacientes con DMD, de acuerdo con la estratificación (3,26,39)
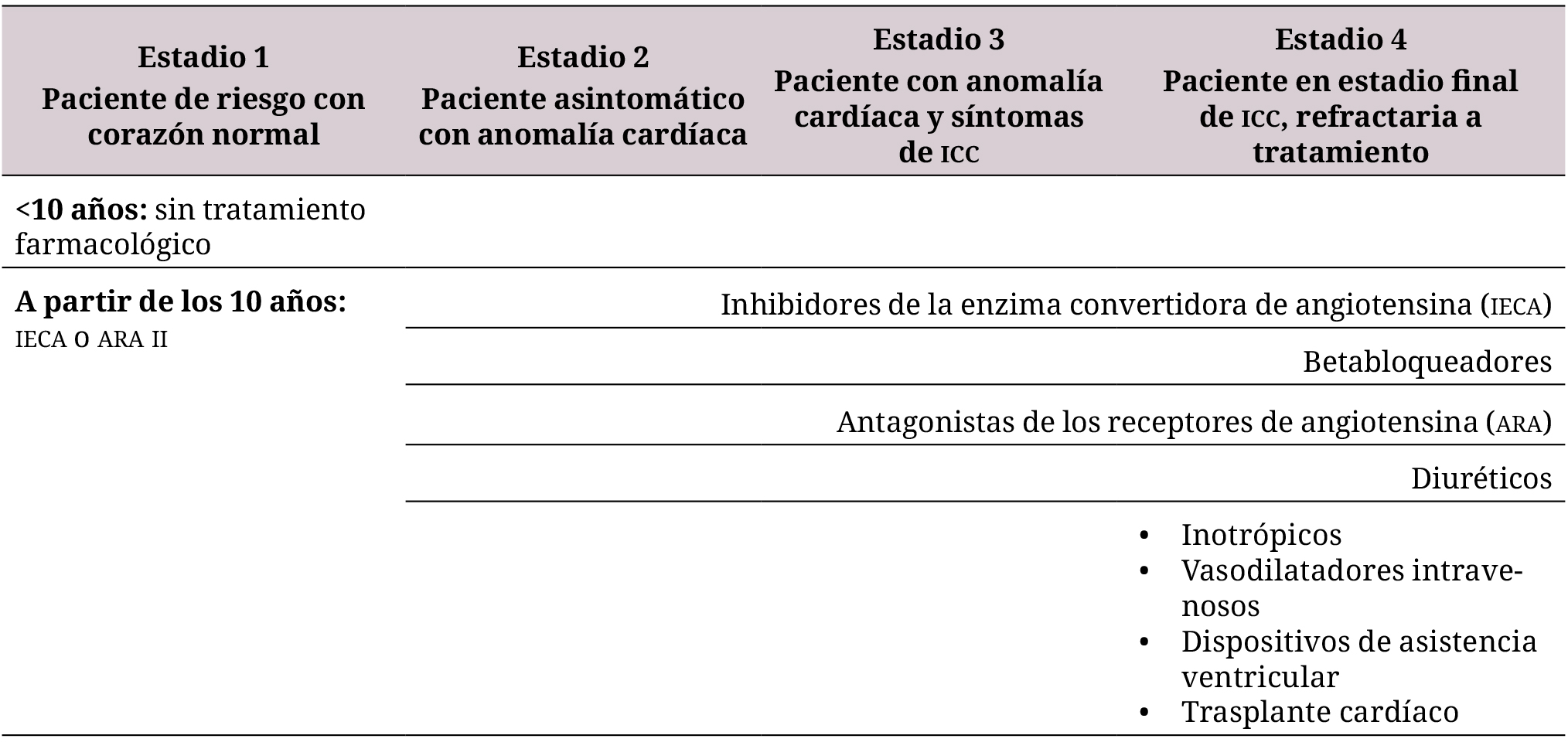
ICC: insuficiencia cardíaca congestiva;
IECA: inhibidores de la enzima convertidora de angiotensina;
ARA: antagonistas de los receptores de angiotensina.
Inhibidores de la enzima convertidora de la angiotensina
Son unos de los medicamentos más utilizados y la primera línea de manejo en pacientes con DMD (1). Los inhibidores de la enzima convertidora de angiotensina (IECA) se usan para enlentecer la remodelación cardíaca que precede al daño del cardiomiocito. Disminuyen la resistencia vascular periférica, mejoran la función endotelial y retrasan la poscarga (1,38).
Las guías actuales recomiendan iniciarlos en pacientes asintomáticos a los 10 años o antes, según la evolución (38,40). Lo anterior se basa en la capacidad de los IECA de retrasar la progresión de la enfermedad cardíaca en la DMD con fracción de eyección normal, mejorando así la sobrevida y calidad de vida de los pacientes (12,23,41).
Inhibidores del receptor de la angiotensina
Los inhibidores del receptor de la angiotensina son la alternativa al uso de IECA, según el tipo de paciente y su edad (1,5,38). Tienen la habilidad de prevenir el daño del cardiomiocito en momentos de esfuerzo cardíaco (38).
Betabloqueadores
Los betabloqueadores se utilizan en combinación con los IECA y usualmente se indican en pacientes con taquicardia o arritmias (38,39), dada la disfunción autonómica en el corazón distrófico de los pacientes con DMD (35). Los estudios de los betabloqueadores como monoterapia en DMD son controversiales, con poca evidencia de lograr efectos positivos importantes (38). Sin embargo, su uso es avalado por la Sociedad Europea de Cardiología, como manejo concomitante con IECA, en pacientes con disfunción sistólica, pues reducen la mortalidad y la tasa de hospitalización (18).
Otras moléculas
La Sociedad Española de Cardiología recomienda el inicio de diuréticos en caso de sobrecarga de líquidos en pacientes sintomáticos o en pacientes refractarios al tratamiento (40). Dentro de los diuréticos, el más usado es la furosemida, como primera línea, y los diuréticos tiazídicos, como medicamentos de segunda línea (40).
En el manejo de la insuficiencia cardíaca en pacientes con DMD, las dosis de los tales medicamentos son similares a las administradas a otros pacientes con insuficiencia cardíaca y miocardiopatía no secundaria a DMD (tabla 5) (3,26,39).
Tabla 5. Dosis de medicamentos para el manejo de la insuficiencia cardíaca en pacientes con distrofia muscular de Duchenne (3,26,39)
IECA |
Betabloqueadores |
Diuréticos/antagonistas de la aldosterona |
Enalapril: |
Carvedilol: |
Furosemida: |
Captopril: |
Metoprolol: |
Espironolactona: |
IECA: inhibidores de la enzima convertidora de angiotensina.
Algunos estudios sugieren que los antagonistas de receptores de mineralocorticoides, por ejemplo, la eplerenona, son eficaces para atenuar la disminución de la función sistólica del ventrículo izquierdo (42,43). Cabe resaltar que la disponibilidad y uso de tratamientos específicos para los problemas cardíacos es de vital importancia en estos pacientes.
Conclusiones
El diagnóstico y el tratamiento precoz de la DMD y la cardiomiopatía, al igual que el seguimiento estrecho y el manejo integral, óptimo y multidisciplinario, son fundamentales para mejorar la calidad de vida y maximizar la supervivencia de los pacientes.
Es ideal realizar una evaluación cardiovascular integral en el momento del diagnóstico de DMD que incluya la valoración clínica por cardiología pediátrica o de adultos, electrocardiograma, ecocardiografía y resonancia magnética nuclear cardíaca. Posteriormente, se debe hacer un seguimiento mediante evaluación cardíaca integral que incluya electrocardiograma, ecocardiograma y biomarcadores. Esta evaluación debe ser anual para los pacientes asintomáticos y cada 3 a 6 meses cuando aparecen alteraciones morfológicas o funcionales.
Se recomienda administrar un IECA o un antagonista de los receptores de la angiotensina en los niños con dmd, comenzando a los 10 años, o antes, según su evolución, para retrasar la progresión de la enfermedad cardíaca.
La insuficiencia cardíaca manifiesta en pacientes con DMD y se trata de acuerdo con las guías de manejo de falla cardíaca.
Contribución de los autores
Todos los autores participaron en la concepción, el diseño, la interpretación de la información, la planeación del artículo, su revisión y aprobaron la versión final del manuscrito.
Coordinación editorial
Integralis HGS (Daniel Rodríguez, MD. y María Stella Salazar, MD.)
Financiación
PTC Therapeutics ha financiado el servicio de medical writing para este artículo.
Conflicto de intereses
Los autores declaran haber participado en el grupo de consenso de manejo de dmd con el apoyo de PTC Therapeutics.
Referencias
1. Fayssoil A, Nardi O, Orlikowski D, Annane D. Cardiomyopathy in Duchenne muscular dystrophy: pathogenesis and therapeutics. Heart Fail Rev. 2010;15(1):103-7.
2. Nigro G, Comi LI, Politano L, Bain RJI. The incidence and evolution of cardiomyopathy in Duchenne muscular dystrophy. Int J Cardiol. 1990;26(3):271-7.
3. Fayssoil A, Abasse S, Silverston K. Cardiac involvement classification and therapeutic management in patients with Duchenne muscular dystrophy. J Neuromuscul Dis. 2017;4(1):17-23.
4. D'Amario D, Amodeo A, Adorisio R, Tiziano FD, Leone AM, Perri G, et al. A current approach to heart failure in Duchenne muscular dystrophy. Heart. 2017 Nov 1;103(22):1770-79.
5. Kaspar RW, Allen HD, Montanaro F. Current understanding and management of dilated cardiomyopathy in Duchenne and Becker muscular dystrophy. J Am Acad Nurse Pract. 2009;21(5):241-9.
6. Cowen L, Mancini M, Martin A, Lucas A, Donovan JM. Variability and trends in corticosteroid use by male United States participants with Duchenne muscular dystrophy in the Duchenne Registry. BMC Neurol. 2019;19(1):1-10.
7. Rafael-Fortney JA, Chadwick JA, Raman SV. Duchenne muscular dystrophy mice and men: can understanding a genetic cardiomyopathy inform treatment of other myocardial diseases? Circ Res. 2016;118(7):1059-61.
8. Adorisio R, Mencarelli E, Cantarutti N, Calvieri C, Amato L, Cicenia M, et al. Duchenne dilated cardiomyopathy: Cardiac management from prevention to advanced cardiovascular therapies. J Clin Med. 2020;9(10):1-18.
9. Gao Q, McNally EM. The dystrophin complex: structure, function and implications for therapy. Compr Physiol. 2015 Jul 1;5(3):1223-39. https://doi.org/10.1002/cphy.c140048
10. Blake DJ, Weir A, Newey SE, Davies KE. Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol Rev. 2002 Apr;82(2):291-329.
11. Goodwin FC, Muntoni F. Cardiac involvement in muscular dystrophies: molecular mechanisms. Muscle Nerve. 2005 Nov;32(5):577-88.
12. Loboda A, Dulak J. Muscle and cardiac therapeutic strategies for Duchenne muscular dystrophy: past, present, and future. Pharmacol Rep. 2020;72:1227-63. https://doi.org/10.1007/s43440-020-00134-x
13. Statile CJ, Taylor MD, Mazur W, Cripe LH, King E, Pratt J, et al. Left ventricular noncompaction in Duchenne muscular dystrophy. J Cardiovasc Magn Reson. 2013;15(1):1.
14. Ashwath ML, Jacobs IB, Crowe CA, Ashwath RC, Super DM, Bahler RC. Left Ventricular dysfunction in Duchenne muscular dystrophy and genotype. Am J Cardiol. 2014 Jul 15;114(2):284-9. https://doi.org/10.1016/j.amjcard.2014.04.038
15. Cripe L, Hor K, Taylor M, Goldberg P, Garrison A, Spicer R, et al. P.2.17 Hypertrophic cardiomyopathy in a patient with Duchenne muscular dystrophy. Neuromuscul Disord. 2013;23(9):754.
16. Tandon A, Taylor MD, Cripe LH. Co-occurring Duchenne muscular dystrophy and hypertrophic cardiomyopathy in an adult with atypical cardiac phenotype. Cardiol Young. 2015 Feb;25(2):355-7.
17. Kim J, Jung IY, Kim SJ, Lee JY, Park SK, Shin HI, et al. A new functional scale and Ambulatory Functional Classification of Duchenne muscular dystrophy: scale development and preliminary analyses of reliability and validity. Ann Rehabil Med. 2018;42(5):690-701.
18. Task A, Members F, Humbert M, Germany MMH, Berger RMF, Denmark JC, et al. 2022 esc/ers guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Heart J. 2022 Oct 11;43(38):3618-3731. https://doi.org/10.1093/eurheartj/ehac237
19. New York Heart Association (nyha). Classification (v2022A) [internet]. [Citado 2022 sep 29]. Disponible en: https://manual.jointcommission.org/releases/TJC2022A/DataElem0439.html
20. Heidenreich PA, Bozkurt B, Aguilar D, Allen LA, Byun JJ, Colvin MM, et al. 2022 aha/acc/ hfsa guideline for the management of heart failure: a report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation. 2022;145:895-1032.
21. Ross RD. The Ross classification for heart failure in children after 25 years: a review and an age-stratified revision. Pediatr Cardiol. 2012 Dec;33(8):1295-300.
22. Hsu DT, Pearson GD. Heart failure in children part I: history, etiology, and pathophysiology. Circ Heart Fail. 2009;2(1):63-70. https://doi.org/10.1161/CIRCHEARTFAILURE.108.820217
23. Price JF. Congestive heart failure in children. Pediatr Rev. 2019;40(2):60-70.
24. Kantor PF, Mertens LL. Clinical practice: heart failure in children. Part I: clinical evaluation, diagnostic testing, and initial medical management. Eur J Pediatr. 2010;169(3):269-79.
25. Buddhe S, Cripe L, Friedland-Little J, Kertesz N, Eghtesady P, Finder J, et al. Cardiac management of the patient with Duchenne muscular dystrophy. Pediatrics. 2018 Oct;142(suppl 2):S72-81. https://doi.org/10.1542/peds.2018-0333I
26. Adorisio R, Mencarelli E, Cantarutti N, Calvieri C, Amato L, Cicenia M, et al. Duchenne dilated cardiomyopathy: cardiac management from prevention to advanced cardiovascular therapies. J Clin Med. 2020;9(10):1-18.
27. Birnkrant DJ, Bushby K, Bann CM, Alman BA, Apkon SD, Blackwell A, et al. Diagnosis and management of Duchenne muscular dystrophy, part 2: respiratory, cardíac, bone health, and orthopaedic management. Lancet Neurol. 2018;17(4):347-61.
28. Nascimento Osorio A, Medina Cantillo J, Camacho Salas A, Madruga Garrido M, Vilchez Padilla JJ. Consensus on the diagnosis, treatment and follow-up of patients with Duchenne muscular dystrophy. Neurologia. 2019;34(7):469-81.
29. Dandel M, Lehmkuhl H, Knosalla C, Suramelashvili N, Hetzer R. Strain and strain rate imaging by echocardiography - basic concepts and clinical applicability. Curr Cardiol Rev. 2009;5(2):133-48.
30. Miyazaki T, Tatara K, Mori K, Inoue M, Hayabuchi Y, Kagami S. Segmental myocardial strain of the left ventricle in patients with Duchenne muscular dystrophy using two-dimensional speckle tracking echocardiography. J Echocardiogr. 2008;6(4):100-8.
31. Spurney CF. Cardiomyopathy of Duchenne muscular dystrophy: current understanding and future directions. Muscle Nerve. 2011;44(1):8-19.
32. Spurney CF, Ascheim D, Charnas L, Cripe L, Hor K, King N, et al. Current state of cardiac troponin testing in Duchenne muscular dystrophy cardiomyopathy: review and recommendations from the Parent Project Muscular Dystrophy expert panel. Open Heart. 2021 Mar;8(1):e001592. https://doi.org/10.1136/openhrt-2021-001592
33. Screever EM, Kootstra-Ros JE, Doorn J, Nieuwenhuis JA, Meulenbelt HEJ, Meijers WC, et al. Kidney function in patients with neuromuscular disease: creatinine versus cystatin C. Front Neurol. 2021 Sep 24;12:688246. https://doi.org/10.3389/fneur.2021.688246
34. Birnkrant DJ, Bushby K, Bann CM, Apkon SD, Blackwell A, Brumbaugh D, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol. 2018;17(3):251-67.
35. Rajdev A, Groh W. Arrhythmias in the muscular dystrophies. Physiol Card Electrophysiol Clin. 2015;7(2):303-8.
36. van Bockel E a. P, Lind JS, Zijlstra JG, Wijkstra PJ, Meijer PM, van den Berg MP, et al. Cardiac assessment of patients with late stage Duchenne muscular dystrophy. Neth Heart J. 2009 Jun;17(6):232-7. https://doi.org/10.1007/BF03086253
37. Spurney CF, McCaffrey FM, Cnaan A, Morgenroth LP, Ghelani SJ, Gordish-Dressman H, et al. Feasibility and reproducibility of echocardiographic measures in children with muscular dystrophies. J Am Soc Echocardiogr. 2015 Aug;28(8):999-1008. https://doi.org/10.1016/j.echo.2015.03.003
38. Meyers TA, Townsend DW. Cardiac pathophysiology and the future of cardiac therapies in Duchenne muscular dystrophy. Int J Mol Sci. 2019;20(17):4098. https://doi.org/10.3390/ijms20174098.
39. Feingold B, Mahle WT, Auerbach S, Clemens P, Domenighetti AA, Jefferies JL, et al. Management of cardiac involvement associated with neuromuscular diseases: a scientific statement from the American Heart Association. 2017 Sep 26;136(13):e200-e231. https://doi.org/10.1161/CIR.0000000000000526
40. Sociedad Española de Cardiología Pediátrica y Cardiopatías Congénitas. Seguimiento cardiológico de la distrofia muscular de Duchenne [internet]. Madrid: Lúa Ediciones; 2021. Disponible en: https://secardioped.org/wp-content/uploads/2021/03/guia-duchenne-sin-logo.pdf
41. Bourke JP, Bueser T, Quinlivan R. Interventions for preventing and treating cardiac complications in Duchenne and Becker muscular dystrophy and X-linked dilated cardiomyopathy. Cochrane Database Syst Rev. 2018;10(10):CD009068. https://doi.org/10.1002/14651858.CD009068.pub3
42. Raman SV, Hor KN, Mazur W, Halnon NJ, Kissel JT, He X, et al. Eplerenone for early cardiomyopathy in Duchenne muscular dystrophy: a randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2015 Feb;14(2):153-61.
43. Raman SV, Hor KN, Mazur W, He X, Kissel JT, Smart S, et al. Eplerenone for early cardiomyopathy in Duchenne muscular dystrophy: results of a two-year open-label extension trial. Orphanet J Rare Dis. 2017 Feb 20;12(1):39.
![]()